Traditional vs. Rapid Microbiological Methods: A 2025 Comparative Analysis for Contamination Detection in Pharma
This article provides a comprehensive comparative analysis for researchers, scientists, and drug development professionals on traditional culture-based and rapid microbiological methods (RMMs).
Traditional vs. Rapid Microbiological Methods: A 2025 Comparative Analysis for Contamination Detection in Pharma
Abstract
This article provides a comprehensive comparative analysis for researchers, scientists, and drug development professionals on traditional culture-based and rapid microbiological methods (RMMs). It explores the foundational principles, advantages, and limitations of each approach, detailing specific methodological applications and technological advancements like PCR, biosensors, and AI. The content addresses critical troubleshooting aspects, including overcoming sampling limitations and detection challenges in complex matrices, and outlines the rigorous validation frameworks required by standards such as the ISO 16140 series and USP chapters <1223> and <1113>. By synthesizing current data and expert consensus, this analysis aims to guide informed method selection to enhance contamination control, accelerate product release, and strengthen quality assurance in pharmaceutical development.
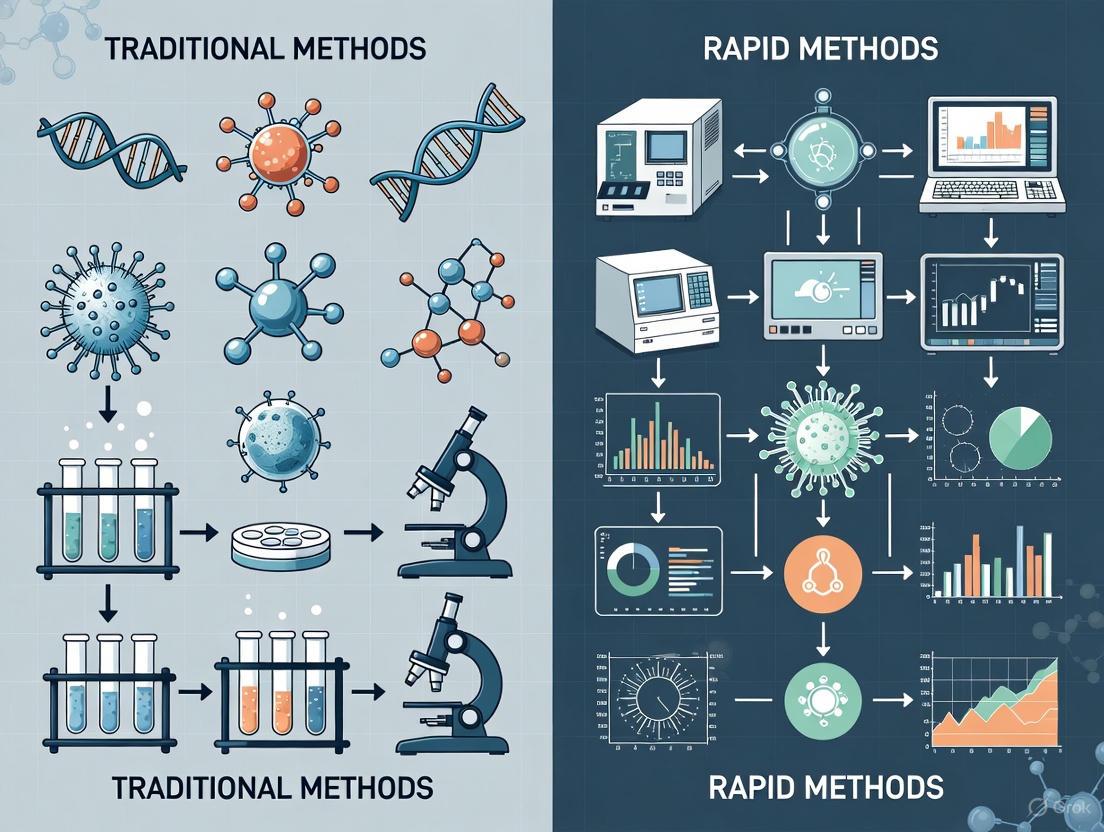
The Microbiological Detection Landscape: From Classic Cultures to Rapid Results
Traditional culture methods remain the foundational approach in diagnostic microbiology for detecting and identifying viable microorganisms. Despite the emergence of rapid technologies, these methods are often considered the "gold standard" due to their proven reliability, ability to detect a wide spectrum of organisms, and provision of live isolates for further analysis [1] [2]. This guide objectively outlines the principles, workflow, and performance of traditional culture methods within the context of contamination detection research.
Core Principles and Relevance
Traditional microbial culture is based on a few fundamental principles. It involves inoculating a sample onto or into nutrient-rich media and incubating it under controlled conditions to support the growth of viable microorganisms [2]. The resulting colonies are then identified based on morphological characteristics, biochemical tests, and microscopic analysis [2].
These methods are highly regarded because they offer a direct and unambiguous confirmation of viable pathogens. They allow for comprehensive antimicrobial susceptibility testing (AST), which is critical for guiding effective treatment and monitoring resistance patterns [3] [2]. Furthermore, the isolation of a pure culture enables detailed genetic and phenotypic studies of the microorganism [4].
The Traditional Culture Method Workflow
The following diagram provides a visual overview of the standard step-by-step workflow for traditional culture methods, from sample collection to final reporting.
Performance Data: Traditional vs. Rapid Methods
The tables below summarize experimental data comparing traditional culture methods with alternative techniques across key performance metrics.
Table 1: Method Comparison in Blood Culture Identification This study compared a rapid centrifugation/Gram stain method with routine culture processing for 152 positive blood culture samples [5].
| Performance Metric | Rapid Centrifugation Method | Routine Culture Method | Agreement |
|---|---|---|---|
| Correct Identification via Gram Stain | 92% (138/150 samples) | Gold Standard | 92% |
| Antibiotic Susceptibility Agreement | N/A | N/A | 97.4% (1934/1984 assays) |
| Time to Preliminary Result | <12 hours | 18-24 hours | N/A |
Table 2: Surface Sampling Method Efficacy for Coliform Detection A laboratory study compared the minimum detection limits of various surface sampling techniques on stainless steel [6].
| Sampling Method | Minimum Detection on Wet Surfaces (cfu/cm²) | Minimum Detection on Dry Surfaces (cfu/cm²) |
|---|---|---|
| Sampling Sponge | ~100 | Markedly Reduced |
| Traditional Hygiene Swabs | <3.5 | Markedly Reduced (less reduction than sponge) |
| Dipslides | <3.5 | Markedly Reduced (less reduction than sponge) |
Detailed Experimental Protocols
Protocol for Surface Sampling and Coliform Detection
This protocol is adapted from a study comparing surface sampling methods [6].
- Objective: To detect and enumerate coliform bacteria on food contact surfaces.
- Materials: Stainless steel coupons, bacterial inoculum, sterile swabs or dipslides, neutralizer solution, selective media (e.g., MacConkey agar).
- Procedure:
- Surface Inoculation: Artificially contaminate sterile stainless steel surfaces with a known concentration of coliform bacteria (e.g., E. coli).
- Drying: Allow surfaces to air-dry for 1 hour in a biosafety cabinet to simulate real-world conditions.
- Sampling: Use standardized technique to sample a defined area (e.g., 10 cm²) with a pre-moistened swab or dipslide.
- Elution: Transfer the sample from the swab into a neutralizer solution to inactivate any disinfectants and vortex thoroughly.
- Culture: Inoculate the eluent onto selective agar plates.
- Incubation & Counting: Incubate plates at 37°C for 24-48 hours and count characteristic colonies to calculate cfu/cm².
Protocol for Blood Culture Processing and AST
This protocol outlines the standard routine method used as a comparator in rapid method evaluations [5].
- Objective: To identify microorganisms and determine their antibiotic susceptibility from a positive blood culture.
- Materials: Automated blood culture system (e.g., Bactec), sheep blood agar (SBA), eosin methylene blue (EMB) agar, biochemical identification panels, AST panels or discs.
- Procedure:
- Subculture: Upon a positive signal from the automated system, aseptically inoculate the blood culture broth onto SBA and EMB agar.
- Primary Incubation: Incubate agar plates at 37°C for 18-24 hours.
- Colony Analysis: Examine for growth and select distinct colonies for Gram staining and further testing.
- Identification: Prepare a standardized suspension from pure colonies (e.g., 0.5 McFarland standard) for biochemical identification using an automated system (e.g., Phoenix) or manual tests.
- AST: Using the standardized suspension, perform AST via disc diffusion, gradient diffusion (Etest), or broth microdilution according to CLSI/EUCAST guidelines [3].
- Interpretation: After 16-24 hours of incubation, measure zones of inhibition or determine Minimum Inhibitory Concentrations (MICs) and interpret as Susceptible (S), Intermediate (I), or Resistant (R) [3].
Essential Research Reagent Solutions
Table 3: Key Materials and Reagents for Traditional Culture Methods
| Item | Function / Explanation |
|---|---|
| Sheep Blood Agar (SBA) | A general-purpose, non-selective medium that supports the growth of a wide range of bacteria and allows for the observation of hemolytic patterns [5]. |
| Selective Media (e.g., EMB, MacConkey Agar) | Contains dyes or inhibitors that suppress the growth of certain bacteria while allowing others to grow, aiding in the preliminary identification of Gram-negative rods like coliforms [5] [6]. |
| Biochemical Identification Panels | Multi-test systems (manual or automated) that determine an organism's metabolic profile, which is then used to identify the species [5]. |
| Mueller-Hinton Agar (MHA) | The standard medium specified by CLSI and EUCAST for performing antimicrobial susceptibility testing via disc diffusion, ensuring reproducible results [3]. |
| McFarland Standards | A reference scale used to standardize the turbidity (and thus the approximate cell density) of bacterial suspensions prior to AST or identification, which is critical for accuracy [5]. |
| Gram Stain Reagents | A fundamental staining procedure that categorizes bacteria into Gram-positive (purple) or Gram-negative (pink) based on their cell wall structure, guiding subsequent testing [5] [2]. |
Advantages and Limitations in Context
Traditional culture methods offer several key advantages: they are well-established and widely accepted by regulatory bodies like the FDA and EPA, provide proven accuracy over a long history of use, and can detect a broad spectrum of bacteria, fungi, and yeast from a single sample [1]. A significant benefit is the ability to obtain a live isolate, which is essential for conducting AST, genetic studies, and epidemiological typing [4].
The primary limitation is the extended time-to-result, typically requiring 24 to 72 hours or more for conclusive identification and AST [1] [3]. The methods are also labor-intensive, requiring significant manual work for media preparation, inoculation, and interpretation, which can introduce human error [1]. Furthermore, they can have limited sensitivity for detecting low levels of contamination or slow-growing organisms that may be outcompeted by faster-growing flora [1].
For decades, the gold standard for microbial detection in pharmaceutical, food, and clinical settings relied heavily on traditional culture-based methods. These techniques, while established and reliable, typically require 48 to 72 hours—or even up to 14 days for sterility testing—to yield results due to their dependence on microbial growth [1] [7]. This significant time delay presents critical challenges for industries requiring rapid product release or timely clinical diagnostics. In response to these limitations, Rapid Microbiological Methods (RMMs) have emerged as transformative technologies that fundamentally reduce detection times, often providing results within hours rather than days [1] [8]. The driving force behind the adoption of these methods is the compelling need for faster time-to-result, which enables quicker decision-making in manufacturing processes, reduces inventory holding costs, and facilitates earlier implementation of corrective actions during contamination events [8].
The evolution from traditional to rapid methods represents more than just an acceleration of testing timelines; it constitutes a fundamental shift in detection philosophy. While traditional methods rely on cultivating microorganisms until they form visible colonies, rapid methods employ sophisticated technologies that detect microbial presence through biomarkers, genetic signatures, or metabolic activities often imperceptible to the human eye [7] [8]. This paradigm shift offers industries unprecedented opportunities to enhance product safety, improve process control, and ultimately better protect public health through more timely and sensitive contamination detection.
Core Rapid Method Technologies: Principles and Applications
Rapid microbiological methods encompass a diverse range of technologies that can be categorized by their underlying detection principles. Each technology offers distinct advantages and is suited to particular applications within the drug development and manufacturing workflow.
Growth-Based Technologies
Growth-based RMMs represent a bridge between traditional and rapid approaches, utilizing conventional liquid or agar media but employing advanced detection systems to identify microbial growth much earlier than visual observation.
Impedance Microbiology: This technique monitors changes in electrical conductivity within growth media resulting from microbial metabolism. As microorganisms grow, they convert electrically neutral substrates into highly charged metabolites (e.g., proteins into amino acids), altering the medium's impedance. These systems can detect changes in conductance (movement of ions between electrodes) or capacitance (charge storage at electrode surfaces), typically detecting ~100,000 CFU for bacteria and ~10,000 CFU for yeast and mold—significantly lower thresholds than turbidity visible to the naked eye [9].
CO₂ Detection Systems: Microbial metabolism in liquid culture produces carbon dioxide, which can be monitored as a viability indicator in closed containers. Systems like the BD BACTEC FX and bioMérieux BacT/ALERT utilize colorimetric or fluorometric sensors that respond to CO₂ accumulation. When CO₂ diffuses into the sensor, it interacts chemically to produce a color change (e.g., from gray to yellow) or fluorescent signal, which the system automatically monitors. The time to detection depends on the initial microbial concentration, with higher loads producing faster responses. This technology has reduced sterility testing for some cell-based products from 14 days to just three days [9].
Viability-Based and Cellular Component Technologies
These systems detect microorganisms through viability stains, cellular components, or metabolic markers without requiring extensive cellular growth, significantly reducing detection times.
Adenosine Triphosphate (ATP) Bioluminescence: This method exploits the nearly universal presence of ATP in living cells. The Celsis Advance II system utilizes the luciferin-luciferase enzyme reaction, which produces light in the presence of ATP. The emitted light is measured by a luminometer, with intensity proportional to the microbial biomass. Some advanced systems incorporate enzyme amplification (e.g., adenylate kinase) to catalyze the conversion of ADP to ATP, enhancing sensitivity beyond native ATP levels alone [10].
Digital Imaging and Autofluorescence: This innovative approach capitalizes on the natural autofluorescence of microbial cells when illuminated with blue light, a property derived from ubiquitous fluorescent biomolecules including flavins, riboflavins, and flavoproteins. Systems utilizing this technology incubate samples on agar cassettes and employ laser excitation coupled with CCD imaging to enumerate micro-colonies in approximately half the time required for visual colony observation [9].
Nucleic Acid-Based Technologies
These methods target genetic material for microbial identification and detection, offering exceptional specificity and sensitivity.
Polymerase Chain Reaction (PCR): The Hygiena BAX System exemplifies PCR-based detection, amplifying specific DNA sequences unique to target organisms. After sample enrichment to increase microbial numbers, cells are lysed to release DNA, which is added to reaction tubes containing primers, nucleotides, and DNA polymerase. Through thermal cycling, target DNA sequences are exponentially amplified, with detection achieved using fluorescent dyes or probes. Results are typically available within 1.5-2.5 hours post-enrichment, offering high specificity for pathogens like Salmonella, Listeria monocytogenes, and E. coli O157:H7 [10].
Matrix-Assisted Laser Desorption/Ionization Time-of-Flight Mass Spectrometry (MALDI-TOF MS): This technology identifies microorganisms based on their unique protein profiles or "fingerprints." Isolated colonies are transferred to a target plate, mixed with a chemical matrix, and irradiated with a laser. The resulting ionized proteins are accelerated through an electric field, with their time-of-flight measured to create a mass spectrum that is compared against an extensive database for identification. This method provides results in minutes rather than the hours or days required for biochemical identification [5] [7].
Table 1: Comparative Analysis of Major Rapid Microbial Detection Technologies
| Technology Category | Example Systems | Detection Principle | Time to Result | Primary Applications | Sensitivity |
|---|---|---|---|---|---|
| Growth-Based (CO₂ Detection) | BD BACTEC FX, bioMérieux BacT/ALERT | Detection of CO₂ production via colorimetric/fluorometric sensors | 8-48 hours [10] | Sterility testing, blood cultures, microbial growth detection | 1 CFU after enrichment [10] |
| Growth-Based (Impedance) | Various systems | Measurement of electrical impedance changes in media due to metabolism | 4-20 hours [10] | Preservative effectiveness testing, microbial screening | ~100,000 CFU (bacteria), ~10,000 CFU (yeast/mold) [9] |
| ATP Bioluminescence | Celsis Advance II, Celsis Accel | Luciferin-luciferase reaction with microbial ATP | 30 min - 48 hours (varies by application) [10] | Bioburden testing, water monitoring, sterility testing | 1 CFU in pre-enriched sample [10] |
| Nucleic Acid-Based (PCR) | Hygiena BAX System, Applied Biosystems MycoSEQ | Amplification of target DNA sequences | 1.5-5 hours [10] | Pathogen detection, mycoplasma testing | <10 CFU or copy equivalent/mL [10] |
| Automated Identification (MALDI-TOF MS) | Bruker MALDI Biotyper | Protein profile fingerprinting by mass spectrometry | Minutes after colony isolation [5] | Microbial identification to genus/species level | Score ≥2.0 indicates species-level identification [5] |
Experimental Protocols and Methodologies
Implementing rapid microbiological methods requires understanding their specific procedural workflows and technical requirements. The following section details experimental protocols for key RMM technologies.
Growth-Based CO₂ Detection Protocol for Sterility Testing
Principle: This method detects microbial growth through the production of carbon dioxide, which is monitored via colorimetric or fluorometric sensors in closed culture vessels [9].
Materials:
- Automated CO₂ detection system (e.g., BD BACTEC FX or bioMérieux BacT/ALERT)
- Appropriate culture bottles with integrated sensors
- Biological safety cabinet
- Positive and negative control strains
Procedure:
- Sample Preparation: Aseptically transfer the test sample (1-10 mL or grams, depending on product type) into culture bottles containing liquid growth media under a biological safety cabinet [10].
- Loading and Incubation: Place inoculated bottles into the automated detection system, which maintains appropriate incubation temperature (typically 30-35°C for mesophiles) [10].
- Monitoring: The system automatically monitors each bottle every 10 minutes for CO₂ production. For colorimetric systems, CO₂ diffusion into the sensor causes a pH decrease, changing the sensor color from gray to yellow. For fluorometric systems, CO₂ production quenches fluorescence [9] [10].
- Result Interpretation: The system algorithms analyze sensor data and flag bottles as positive when CO₂ production exceeds predetermined thresholds. Positive bottles typically require subculturing to confirm microbial growth and for identification [9].
- Final Reading: Negative bottles are typically incubated for 5-7 days (depending on methodology) before final determination [7].
Validation Parameters: Include accuracy, precision, limit of detection, robustness, and equivalence to compendial methods as per USP <1223> and FDA guidelines [7].
Nucleic Acid-Based Detection Protocol Using Real-Time PCR
Principle: This method detects and identifies microorganisms by amplifying specific DNA sequences unique to target organisms using fluorescently monitored PCR [10] [11].
Materials:
- Real-time PCR instrument (e.g., thermal cycler with fluorescence detection capability)
- Species-specific primers and probes
- DNA extraction reagents (lysis buffers, enzymatic reagents, purification columns/magnetic beads)
- Positive and negative control DNA templates
- PCR master mix (containing DNA polymerase, dNTPs, buffer)
Procedure:
- Sample Enrichment: Incubate sample in appropriate growth media to increase microbial biomass and ensure detection of viable organisms. Typical enrichment times range from 18-24 hours [10].
- DNA Extraction: a. Transfer 1 mL of enriched sample to a microcentrifuge tube. b. Centrifuge at 10,000-14,000 × g for 2 minutes to pellet cells. c. Resuspend pellet in enzymatic lysis solution (e.g., containing lysozyme for Gram-positive bacteria) and incubate at appropriate temperature. d. Purify DNA using magnetic beads or silica-based columns according to manufacturer's instructions. e. Elute DNA in appropriate buffer (e.g., TE buffer or nuclease-free water) [10].
- PCR Setup:
a. Prepare reaction mixture containing:
- 12.5 μL PCR master mix
- 2.5 μL primer-probe mix (containing target-specific primers and fluorescent probe)
- 5 μL template DNA
- Nuclease-free water to 25 μL total volume b. Load reactions into real-time PCR instrument [10].
- Amplification and Detection:
a. Program thermal cycler with appropriate protocol:
- Initial denaturation: 95°C for 2-10 minutes
- 40-45 cycles of:
- Denaturation: 95°C for 15-30 seconds
- Annealing/Extension: 60°C for 30-60 seconds (with fluorescence acquisition) b. The system monitors fluorescence during each cycle, with a significant increase in fluorescence signal indicating amplification of the target sequence [10].
- Data Analysis: The software calculates cycle threshold (Ct) values for each sample, with lower Ct values indicating higher initial target concentration. Results are interpreted as positive or negative based on predetermined Ct cutoffs [10].
Figure 1: Real-Time PCR Workflow for Microbial Detection
The Scientist's Toolkit: Essential Research Reagent Solutions
Successful implementation of rapid microbiological methods requires specific reagents and materials tailored to each technology platform. The following table details essential research reagent solutions for the featured RMMs.
Table 2: Essential Research Reagent Solutions for Rapid Microbiological Methods
| Reagent/Material | Technology Application | Function | Technical Specifications |
|---|---|---|---|
| Selective Culture Media Vials | Growth-based CO₂ Detection (e.g., BioLumix System) | Supports growth of target microorganisms while inhibiting competitors; contains specific substrates and indicators | Formulated with selective agents (e.g., antibiotics, chemicals), indicators for color/fluorescence change; specific for total aerobes, yeast/mold, coliforms, E. coli, etc. [10] |
| Luciferin-Luciferase Enzyme Reagent | ATP Bioluminescence (e.g., Celsis systems) | Catalyzes the conversion of microbial ATP into light (bioluminescence) | Contains purified luciferase enzyme, luciferin substrate, buffer; must be stored at specified temperatures to maintain activity [10] |
| DNA Extraction Kits | Nucleic Acid-Based Methods (PCR) | Isolates and purifies microbial DNA from samples | Includes lysis buffers (enzymatic/mechanical), protease, binding columns/magnetic beads, wash buffers, elution buffer; designed for efficient recovery of microbial DNA [10] |
| PCR Master Mix | Nucleic Acid-Based Methods (PCR) | Provides essential components for DNA amplification | Contains thermostable DNA polymerase, dNTPs, MgCl₂, reaction buffers; often includes optimized formulations for different sample types [10] |
| MALDI-TOF Matrix Solution | MALDI-TOF Mass Spectrometry | Facilitates sample ionization and desorption for mass spectrometry | Typically α-cyano-4-hydroxycinnamic acid (HCCA) in organic solvent/water mixture with trifluoroacetic acid; must be fresh for optimal crystallization [5] |
| Fluorescent Stains for Micro-Colonies | Viability-Based Detection (e.g., fluorescent staining methods) | Stains microbial cells for laser enumeration | Non-fluorescent substrates taken up by microorganisms and enzymatically cleaved to liberate fluorochrome; accumulates in cell cytoplasm for signal amplification [9] |
| Impedance Microbiology Media | Impedance-Based Detection | Specially formulated for electrical signal detection | Contains substrates that generate charged metabolites during microbial growth; optimized conductivity for specific instrument systems [9] |
Comparative Performance Data: Rapid vs. Traditional Methods
Understanding the relative performance of rapid methods compared to traditional approaches is essential for method selection and validation. The following comparative data highlights the advantages and limitations of each technology.
Table 3: Performance Comparison of Rapid vs. Traditional Microbial Methods
| Performance Characteristic | Traditional Culture Methods | Rapid Microbial Methods | Comparative Advantage |
|---|---|---|---|
| Time to Result (Qualitative) | 24-72 hours [1] | 1.5-24 hours [10] | Up to 94% reduction in detection time [1] [10] |
| Time to Result (Sterility Test) | 14 days [7] | 3-7 days [9] [7] | Up to 79% reduction in testing time [9] |
| Sensitivity | Detects culturable organisms only [12] | Detects viable but non-culturable (VBNC) organisms [12] [8] | Broader detection spectrum including stressed organisms [8] |
| Specificity | Moderate (based on colony morphology) | High (based on genetic, protein, or metabolic signatures) [1] | Reduced false positives/negatives [1] |
| Automation Potential | Low (labor-intensive) [1] | High (automated systems available) [1] [8] | Reduced labor costs and human error [1] |
| Throughput | Low to moderate | Moderate to high (e.g., 96 tests in 1.5-2.5 hours for PCR) [10] | Increased testing capacity [10] |
| Quantitative Capability | Yes (CFU enumeration) | Yes (various units: RLU, gene copies, etc.) [8] | Faster quantification [8] |
| Regulatory Acceptance | Well-established and widely accepted [1] | Increasing acceptance with validation [7] | Requires demonstration of equivalence [7] |
Figure 2: Performance Comparison Between Traditional and Rapid Methods
The landscape of microbiological testing is undergoing a profound transformation driven by technological innovation and the pressing need for faster, more accurate detection methods. Rapid microbiological methods represent a significant advancement over traditional culture-based approaches, offering reduced time-to-result, enhanced sensitivity, and improved automation capabilities. While implementation challenges remain—including initial investment costs, validation requirements, and regulatory acceptance—the compelling benefits of these technologies are accelerating their adoption across pharmaceutical, clinical, and food industries [1] [7] [8].
The future of microbial detection will likely see increased integration of these technologies into quality control systems, with real-time monitoring becoming more prevalent in manufacturing environments. As regulatory guidance continues to evolve and validation frameworks become more standardized, rapid methods will progressively shift from supplemental to primary detection platforms. For researchers and drug development professionals, understanding these technologies' principles, applications, and implementation requirements is essential for leveraging their full potential to enhance product safety and public health protection.
Microbiological testing is fundamental to ensuring product safety and quality across critical industries such as pharmaceuticals, food and beverage, and cosmetics [1]. For decades, traditional culture-based methods were the undisputed standard for detecting and enumerating microorganisms. However, the evolution of technology and increasing demand for faster results have catalyzed the development and adoption of Rapid Microbiological Methods (RMMs), which offer a paradigm shift in testing speed and efficiency [1] [7]. This guide provides an objective, high-level comparison of these two approaches, detailing their respective strengths, weaknesses, and performance characteristics to inform researchers, scientists, and drug development professionals in their method selection and validation processes.
Traditional Microbial Methods
Traditional methods rely on the growth of microorganisms on specific culture media, with incubation times typically ranging from 48 to 72 hours, or even longer for slow-growing organisms [1] [7]. These techniques, such as the plate count method, involve inoculating samples onto petri dishes, incubating them, and subsequently counting Colony Forming Units (CFUs) to estimate the microbial load [1].
Rapid Microbiological Methods (RMMs)
RMMs encompass a suite of advanced technologies—including molecular methods (e.g., PCR, NGS), immunoassays (e.g., ELISA), and sensor-based systems (e.g., biosensors)—that significantly reduce the time to result, often delivering outcomes in a matter of hours or even minutes [1] [13]. These methods are designed to detect microorganisms, their components (like nucleic acids or proteins), or metabolic products without relying solely on lengthy culture steps.
High-Level Comparative Table
Table 1: A high-level comparison of traditional and rapid microbiological methods.
| Feature | Traditional Methods | Rapid Microbiological Methods (RMMs) |
|---|---|---|
| Time to Result | 24-72 hours or longer [1] [7] | As little as a few hours or minutes [1] [14] |
| Level of Automation | Low; predominantly manual and labor-intensive [1] | High; many systems are automated, reducing human error [1] |
| Sensitivity | Can be limited, may miss low-level contamination [1] | High sensitivity and specificity; can detect very low levels of pathogens [1] [14] |
| Scope of Detection | Broad; detects all viable bacteria, fungi, and yeast [1] | Can be targeted; may not be amenable to all microorganisms or samples [1] |
| Regulatory Status | Well-established and widely accepted by regulatory bodies [1] [7] | Gaining relevance but may require additional validation for compliance [1] [7] |
| Initial Investment | Low equipment requirements; highly economical for small labs [1] | High initial setup costs for equipment and training [1] [7] |
| Data Richness | Provides additional information on microbial populations [1] | Provides rapid, specific identification but may offer less cultural data [7] |
| VBNC Detection | Cannot detect Viable But Non-Culturable (VBNC) organisms [7] | Capable of detecting VBNC and stressed organisms [7] |
Experimental Data and Performance Comparison
Validation and Equivalency Data
For an RMM to be adopted in regulated industries, it must demonstrate equivalency to traditional methods. A study on the Sievers Soleil Rapid Bioburden Analyzer, which utilizes a rapid ATP-bioluminescence technology, showed strong performance against traditional membrane filtration [14].
Table 2: Performance data of a Rapid Microbiological Method (RMM) versus traditional plate count [14].
| Performance Metric | Result | Acceptance Criteria |
|---|---|---|
| Linearity (R²) | >0.95 for 3-4 logs of organisms | >0.95 per USP <1223> |
| Accuracy (Avg. % Recovery) | 140.9% | >50% with a goal of <200% |
| Limit of Quantification (LOQ) | 0.1 CFU/mL | Established per validation protocol |
| Limit of Detection (LOD) | 0.05 CFU/mL | Established per validation protocol |
The average percent recovery of 140.9%, while passing the acceptance criteria, indicates a consistent, though slightly elevated, recovery compared to the traditional method, which is considered acceptable for this validation [14].
Performance in Clinical Blood Culture
A 2020 study directly compared a rapid centrifugation and Gram staining method against routine processing for positive blood culture samples, a critical scenario for patient treatment [5]. The rapid method achieved:
- 92% agreement (138 out of 152 samples) with routine procedures for bacterial strain identification [5].
- 97.4% agreement in antibiotic susceptibility testing profiles across 1,984 antibiotic susceptibility assays [5]. This demonstrates that rapid methods can be successfully integrated into time-sensitive clinical workflows without sacrificing accuracy.
Suitability for Product Testing
A study evaluating 3M Petrifilm rapid tests across various dietary supplements showed its suitability as an alternative to gold standard methods [15]. For tests including Total Aerobic Microbial Count (TAMC) and specific pathogen detection, recovery rates compared to the control consistently exceeded 70%, meeting the acceptance criteria set by U.S. Pharmacopeia [15]. Recovery ranges for different product types, such as multivitamins (79%-111%) and protein powders (94%-104%), highlight the robustness of this RMM across diverse matrices [15].
Detailed Experimental Protocols
Protocol: Rapid Identification from Blood Cultures
The following protocol, adapted from a 2020 study, outlines a rapid method for identifying and conducting antibiotic susceptibility tests from positive blood culture vials within the first 12 hours of incubation [5].
1. Sample Collection:
- Draw 5 mL of liquid from a blood culture vial that has signaled positive within 12 hours in an automated incubator (e.g., Bactec FX).
2. Centrifugation:
- Transfer the 5 mL sample into a standard blood collection tube.
- Centrifuge at 2,000 rpm for 10 minutes. This process separates blood components below a gel layer, concentrating bacteria in a film layer on top of the gel.
3. Gram Staining:
- Carefully remove the supernatant liquid.
- Use a cotton swab to collect a sample from the bacterial film layer above the gel.
- Smear the sample onto a microscope slide and air-dry.
- Perform Gram staining sequentially with: crystal violet (2 minutes), Lugol's solution (2 minutes), decolorizer (alcohol), and counterstain (diluted fuchsin, 30 seconds). Wash with water between each step [5].
- Air-dry the slide and examine under an oil immersion lens (100x objective) to categorize organisms as Gram-positive or Gram-negative.
4. Identification and Susceptibility Testing:
- From the gel layer, pick bacteria and suspend them in an identification broth to a density equivalent to a 0.5-0.63 McFarland standard.
- Transfer a portion of this suspension to an antibiotic susceptibility test (AST) broth.
- Load the ID and AST solutions into an automated system (e.g., Phoenix 100) with appropriate test kits.
- Results are typically available in 8-12 hours, allowing for reporting within 24 hours of the initial positive signal [5].
5. Comparative Analysis (Routine Method):
- In parallel, inoculate the positive blood culture sample onto solid media (e.g., Sheep Blood Agar, EMB Agar) without centrifugation.
- Incubate at 37°C for 18-24 hours.
- Perform Gram staining and antibiotic susceptibility testing from the grown colonies the following day, following the same automated procedure [5].
Protocol: Validation of a Rapid Bioburden Method
This protocol is based on a study evaluating the Sievers Soleil Rapid Bioburden Analyzer against the traditional membrane filtration method, following pharmacopeial recommendations [14].
1. Microorganism Selection and Preparation:
- Select microorganisms per USP ⟨1223⟩, EP 5.1.6, and JP G4 guidelines. The panel should include gram-positive and gram-negative bacteria, yeast, and fungi (e.g., B. subtilis, E. coli, P. aeruginosa, C. albicans, A. brasiliensis) [14].
- Include stressed or starved organisms (e.g., starved for three days) to simulate real-world conditions and challenge the method.
- Create a stock solution and perform serial dilutions to achieve test concentrations spanning from 0.05 CFU/mL to 100 CFU/mL.
2. Inoculation and Testing:
- Add the microorganisms to a representative sample matrix, such as Water For Cell Culture.
- Aliquot the inoculated sample for testing in parallel on both the RMM (Sievers Soleil) and the traditional method (membrane filtration followed by plate incubation).
- Include a sufficient number of replicates (e.g., 10 replicates for lower concentrations) and negative controls.
3. Data Analysis and Equivalency Determination:
- For the RMM, results are generated based on microbial detection technology (e.g., bioluminescence).
- For the traditional method, count CFUs after the appropriate incubation period.
- Calculate the recovery of the RMM compared to the traditional method.
- Establish linearity, accuracy, precision, Limit of Detection (LOD), and Limit of Quantification (LOQ) per validation guidelines [14].
- The method is considered equivalent if it meets pre-defined acceptance criteria, such as a recovery of >50% with a goal of <200% and a linearity R² of >0.95 [14].
Workflow Visualization
The following diagram illustrates the key decision-making workflow when choosing between traditional and rapid microbiological methods, highlighting critical considerations at each stage.
Microbiological Method Selection Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
Successful implementation and validation of microbiological methods, whether traditional or rapid, rely on a suite of essential reagents and materials.
Table 3: Essential research reagents and materials for microbiological testing.
| Item | Function | Application Context |
|---|---|---|
| Selective & Non-Selective Culture Media (e.g., SBA, EMB Agar) | Supports the growth and differentiation of microorganisms. | Traditional culture-based methods for isolation and enumeration [5]. |
| Gram Staining Kits (Crystal Violet, Iodine, Decolorizer, Counterstain) | Differentiates bacteria into Gram-positive and Gram-negative based on cell wall structure. | First-line rapid characterization in both traditional and rapid workflows [5]. |
| Identification & Susceptibility Test Kits (e.g., GP/GN ID/AST) | Contains substrates and antibiotics for automated identification and susceptibility testing. | Used with systems like Phoenix 100 for both rapid and traditional method endpoints [5]. |
| MALDI-TOF MS Matrix & Calibration Standards | Enables protein "fingerprinting" of microorganisms for rapid identification. | Rapid Identification RMM; used on colonies from traditional plates or from direct sample processing [5] [7]. |
| PCR Reagents (Primers, Probes, Master Mix) | Amplifies specific microbial DNA/RNA sequences for detection and quantification. | Molecular RMMs (e.g., PCR, qPCR) for highly sensitive and specific pathogen detection [1] [13]. |
| ATP Bioluminescence Reagents (Luciferin/Luciferase) | Produces light in proportion to the amount of microbial ATP present, indicating contamination. | Rapid hygiene monitoring and quantitative RMMs like the Sievers Soleil analyzer [14] [7]. |
The choice between traditional and rapid microbiological methods is not a matter of one being universally superior to the other. Instead, it is a strategic decision that must balance speed, cost, regulatory requirements, and the specific informational needs of the product or research context [1]. Traditional methods remain the robust, gold standard for broad-spectrum analysis where time is not the primary constraint. In contrast, RMMs offer a powerful alternative for time-sensitive applications, high-throughput environments, and when targeting specific pathogens with high sensitivity is critical [1] [13]. The ongoing validation and adoption of RMMs, supported by the experimental data and protocols outlined in this guide, are steadily enhancing our ability to ensure product safety and quality with unprecedented efficiency.
Industry Adoption Trends and Historical Barriers to RMM Implementation
The field of microbiological quality control is undergoing a significant transformation, moving from traditional, slow culture-based methods toward Rapid Microbial Methods (RMMs). Traditional methods, while considered the gold standard for detecting microbial contamination, are labor-intensive and time-consuming, often requiring several days to yield results due to the need for microbial growth [16]. This delay can impact critical decision-making processes in pharmaceutical manufacturing and biopharmaceutical sectors, particularly for products with short shelf lives [17]. In response, RMMs have emerged as powerful tools that offer faster results, increased sensitivity, and improved efficiency [14]. These innovative techniques, which include technologies like polymerase chain reaction (PCR), next-generation sequencing (NGS), and mass spectrometry, are revolutionizing contamination detection by providing real-time or near-real-time data [16] [18].
The adoption of RMMs is not merely a technological upgrade but a strategic imperative driven by the need for proactive risk management and operational excellence. The growing demand for quicker and more accurate microbial testing methods is propelling the market forward, with the automated and rapid microbiological testing market projected to reach $5.89 billion by 2033 [18]. However, despite the clear advantages and overwhelming interest—with 93% of surveyed professionals expressing interest in rapid sterility testing—the adoption rate remains relatively low, with only 28% currently utilizing these methods [17]. This discrepancy highlights the existence of significant barriers that hinder widespread implementation. This article explores the historical adoption trends, identifies key implementation barriers, and provides a comparative analysis of traditional versus rapid methods through experimental data, offering a comprehensive resource for researchers, scientists, and drug development professionals navigating this evolving landscape.
Historical Barriers to Widespread RMM Implementation
The journey toward adopting Rapid Microbial Methods has been fraught with challenges that have slowed their integration into quality control systems. Understanding these barriers is crucial for developing effective strategies to overcome them.
High Initial Investment and Cost Concerns: The adoption of RMMs in pharmaceutical manufacturing often involves significant capital expenditure. Costs are associated not only with the purchase of instruments but also with installation, qualification, and implementation [17]. This substantial upfront investment can be a major deterrent, particularly for smaller organizations with limited capital budgets. A survey of pharmaceutical and biopharmaceutical experts identified the cost of instruments and tests as the top concern when considering implementing RMMs [17].
Validation Complexities and Regulatory Uncertainty: The validation process for rapid platforms often raises concerns among experts due to its perceived complexity compared to traditional methods [17]. Organizations may be uncertain about regulatory acceptance of these new technologies, creating hesitation in adoption. However, it is important to note that regulatory agencies are increasingly accepting of RMMs and actively encourage their adoption. Guidance documents such as the PDA Technical Report #33, United States Pharmacopeia (USP)
<1223>, and the European Pharmacopoeia (EP) chapter 5.1.6 offer direction on validation strategies, which can alleviate many concerns about the validation process [17].Technical and Workforce Readiness Challenges: Another significant barrier is the shortage of qualified microbiologists and laboratory technicians in some regions capable of operating these complex systems [18]. After the initial implementation of a new RMM, staff need time to adjust to new workflows, requiring comprehensive training and change management initiatives. Success depends on fostering trust in the new technologies and adapting to evolving job roles and skill requirements [17] [19].
Perceived Limitations in Accuracy and Sensitivity: While rapid microbiological tests offer quicker results, some organizations question whether their accuracy and sensitivity always match traditional testing methods [18]. This perception of potentially compromised reliability can make organizations reluctant to fully adopt automated testing systems until further advancements demonstrate consistent performance. The industry is awaiting more extensive data and case studies that validate the precision of RMMs across diverse applications.
Integration with Legacy Systems: Many industries still operate with legacy infrastructure, fragmented systems, or outdated IT environments that don't easily connect with modern RMM platforms [19]. This makes integration complex, often requiring custom connectors, retrofitting, and modernization efforts to ensure interoperability and scalability. Overcoming this challenge requires platform modernization, API-driven integration, and process re-engineering to create a seamless technological ecosystem.
Table 1: Summary of Key Barriers to RMM Implementation
| Barrier Category | Specific Challenges | Impact on Adoption |
|---|---|---|
| Economic Factors | High upfront costs for instruments, installation, and qualification; Ongoing maintenance expenses [17] [18] | Significant barrier for SMEs and organizations with limited capital budgets |
| Regulatory Compliance | Perceived complexity of validation process; Evolving regulatory standards; Lack of standardized protocols [17] [18] | Creates uncertainty and slows decision-making; Increases development costs for vendors |
| Technical Expertise | Shortage of qualified personnel; Extensive training requirements; Need for change management [17] [18] | Limits widespread adoption; Increases implementation time and costs |
| Performance Concerns | Questions about accuracy/sensitivity compared to traditional methods; Reliability in complex matrices [18] | Creates reluctance to fully adopt until more validation data is available |
| System Integration | Compatibility with legacy infrastructure; Need for custom connectors; Data interoperability issues [19] | Increases complexity and cost of implementation; May require additional modernization investments |
Current Adoption Trends and Market Trajectory
Despite implementation barriers, the RMM market is experiencing robust growth driven by technological advancements and evolving industry needs. The automated and rapid microbiological testing market was valued at $3.25 billion in 2023 and is projected to reach $5.89 billion by 2033, growing at a Compound Annual Growth Rate (CAGR) of 7.25% between 2026 and 2033 [18]. This growth trajectory underscores the increasing acceptance and integration of these technologies across various sectors.
Several key trends are shaping the current adoption landscape. There is a notable expansion of applications beyond traditional pharmaceutical quality control, including food safety, environmental monitoring, and dairy industry applications [20]. In the realm of food safety, emerging detection technologies are addressing limitations of traditional methods in dealing with complex supply chains and diverse microbial contaminants [16]. The integration of artificial intelligence (AI) and machine learning (ML) with RMM systems is producing more reliable results and enabling predictive analytics [18]. Furthermore, the adoption of cloud-based data management systems is enhancing scalability and accessibility while facilitating real-time monitoring and decision-making [21].
The market is also witnessing increased consolidation and competition, with a mix of established players and innovative startups driving technological advancements [21]. Mergers and acquisitions are common as companies seek to expand their technological capabilities and customer base. The regulatory landscape is simultaneously evolving to accommodate new technologies, particularly for advanced therapy medicinal products (ATMPs) with short shelf lives that benefit significantly from rapid testing methods [17]. These converging trends indicate a promising future for RMM adoption as technological capabilities advance, implementation costs potentially decrease, and regulatory pathways become more clearly defined.
Comparative Analysis: Traditional Methods vs. Rapid Microbial Methods
A comprehensive understanding of the performance differences between traditional and rapid methods is essential for informed decision-making. The following experimental data and comparison tables provide objective insights into the capabilities of modern RMM platforms.
Experimental Protocol for RMM Evaluation
To ensure reliable evaluation of RMMs, rigorous testing protocols must be established. The following methodology outlines a standardized approach for comparing RMM performance against traditional methods:
Microorganism Selection: Microorganisms are chosen according to recommendations from major pharmacopoeias, including United States Pharmacopeia (USP) Chapter
<1223>"Validation of Alternative Microbiological Methods," European Pharmacopoeia Chapter 5.1.6 "Alternative Methods for Control of Microbiological Quality," and Japanese Pharmacopoeia General Information G4 [14]. Test organisms should include both representative ATCC strains and environmental isolates relevant to the specific application. Typical strains include A. brasiliensis, B. subtilis, C. albicans, E. coli, P. aeruginosa, and S. aureus, among others [14].Sample Preparation and Inoculation: Microorganisms are prepared in stock solutions and placed under starving conditions for three days to simulate real-world stress conditions [14]. Serial dilutions are prepared for each stock solution across a range of concentrations (e.g., 0.05, 0.1, 1, 10, and 100 CFU/mL). For each concentration level, multiple replicates are tested (e.g., ten replicates for lower concentrations and six for higher concentrations) to ensure statistical significance [14].
Parallel Testing and Comparison: Samples are tested in parallel using both the RMM platform and traditional methods (e.g., membrane filtration for liquid samples). Negative control samples are included throughout the study to account for potential contamination [14]. System suitability standards at different concentrations are run during daily start-up to ensure consistent instrument performance [14].
Data Analysis and Acceptance Criteria: Key validation parameters include linearity (with a correlation coefficient >0.95 per USP
<1223>), accuracy and precision (recovery compared to agar plates >50% with a goal of <200%), and determination of the Lower Limit of Detection (LLOD) and Lower Limit of Quantification (LLOQ) [14].
Performance Comparison Table
The following table summarizes key performance metrics for traditional methods versus RMMs based on experimental data:
Table 2: Performance Comparison of Traditional Culture Methods vs. RMMs
| Performance Metric | Traditional Culture Methods | Rapid Microbial Methods (e.g., Sievers Soleil) | Data Source |
|---|---|---|---|
| Time to Result (TTR) | 2-7 days (depending on method and microbe) | ≤45 minutes for bioburden results | [14] |
| Limit of Detection (LOD) | Varies by method; typically ~1 CFU/sample | 0.05 CFU/mL | [14] |
| Limit of Quantification (LOQ) | Varies by method; typically ~10 CFU/sample | 0.1 CFU/mL across all test organisms | [14] |
| Accuracy/Recovery | Established as gold standard | 140.9% average recovery (meeting >50% acceptance criteria) | [14] |
| Linearity | Not typically measured for qualitative methods | >0.95 correlation coefficient across 3-4 logs | [14] |
| Automation Capability | Largely manual processes | Fully automated, reducing manual intervention | [17] [18] |
| Labor Requirements | High (manual preparation, incubation, reading) | Low after initial setup and training | [17] |
Technology-Specific Capabilities
Different RMM technologies offer varying strengths and limitations, making them suitable for different applications:
Table 3: Comparison of Rapid Microbial Method Technologies
| Technology | Strengths | Limitations | Best Use Cases |
|---|---|---|---|
| PCR/qPCR | High specificity (100% for target genes); Rapid (2-4 hours) [16] | Limited to known targets; Risk of false negatives in complex matrices [16] | Confirming pathogen presence in processed foods (e.g., Listeria in cheese) [16] |
| MALDI-TOF MS | Species-level identification (>95% accuracy); Minimal sample preparation [16] | High equipment cost; Requires extensive reference databases [16] | Rapid identification of E. coli O157:H7 in meat products [16] |
| Next-Generation Sequencing | Pathogen traceability (SNP resolution); Detects unknown pathogens [16] | High computational cost (>100 GB/sample); Time-intensive (24-72 hours) [16] | Outbreak tracing (e.g., linking Salmonella strains to contaminated poultry farms) [16] |
| Electrochemical Sensors | Portable, real-time detection; Low cost (<$100/device) [16] | Limited multiplexing capability; Calibration drift in field conditions [16] | On-site monitoring of Vibrio in seafood during transportation [16] |
| Automated Viability-Based Systems | Distinguishes between live and dead cells; No complex sample preparation | May have higher instrument costs; Requires specific training | Pharmaceutical manufacturing; Sterility testing |
The Scientist's Toolkit: Essential Research Reagent Solutions
Implementing and validating RMMs requires specific reagents and materials designed to ensure accurate and reproducible results. The following table details key research reagent solutions essential for RMM evaluation and routine use:
Table 4: Essential Research Reagent Solutions for RMM Implementation
| Reagent/Material | Function | Application in RMM Validation |
|---|---|---|
| Pharmacopoeia-Strain Microorganisms | Reference strains for method validation and qualification | Establishing method accuracy, precision, and linearity per regulatory guidelines [14] |
| Environmental Isolates | Real-world microorganisms from manufacturing facilities | Demonstrating method robustness for actual use case scenarios [14] |
| System Suitability Standards | Quality control standards for instrument performance verification | Daily verification of instrument sensitivity and detection capabilities [14] |
| Viability Markers | Chemical indicators of cellular metabolic activity | Differentiating between live and dead cells in viability-based methods |
| Nucleic Acid Extraction Kits | Isolation of DNA/RNA from microbial cells | Preparing samples for molecular-based RMMs (PCR, sequencing) [16] |
| Culture Media & Diluents | Microbial growth support and sample matrix | Method comparison studies against traditional culture methods [14] |
| Positive & Negative Controls | Reference materials for result interpretation | Ensuring test validity and detecting potential contamination [14] |
Future Outlook: Emerging Technologies and Research Directions
The future of RMMs is closely tied to technological advancements and evolving regulatory frameworks. Several emerging trends are poised to further transform the landscape of microbiological testing:
Integration of Artificial Intelligence and Machine Learning: AI and ML are increasingly being incorporated into RMM platforms to enhance data analysis, enable predictive modeling, and improve detection accuracy. These technologies can identify patterns in complex datasets that might be missed by human analysts, potentially reducing false positives and negatives [18]. AI-driven models integrating multi-omics data are already showing improved prediction capabilities, with error rates decreasing from ±1.5 log CFU to ±0.8 log CFU in some applications [16].
Advancements in Molecular Technologies: Emerging detection methods such as Recombinase Polymerase Amplification (RPA), CRISPR-based diagnostics, and viability-based methods (e.g., PMA-qPCR) are gaining traction for their specificity, sensitivity, and rapid turnaround times [20]. These technologies are particularly valuable for detecting specific pathogens in complex matrices and for distinguishing between viable and non-viable microorganisms, a critical distinction in many pharmaceutical and food safety applications.
Miniaturization and Portable Testing Platforms: The development of portable, handheld detection devices is enabling real-time, on-site microbial monitoring, reducing the need for sample transport and centralized laboratory testing [16]. These platforms are particularly valuable for environmental monitoring, supply chain surveillance, and point-of-care testing applications where rapid results are essential for timely decision-making.
Integration with Blockchain and IoT: The combination of RMMs with blockchain technology and Internet of Things (IoT) devices enables real-time monitoring and secure data tracking throughout the supply chain [16]. For example, whole-genome sequencing data combined with blockchain can reduce contamination response time to 48 hours in poultry supply chains, demonstrating how digital technologies can enhance the value of RMMs [16].
Regulatory Evolution and Standardization: Regulatory policies in both the US and the EU are evolving to accommodate the unique requirements of short shelf-life products and the need for rapid testing [17]. This regulatory evolution, coupled with increasing international harmonization of standards, will likely accelerate RMM adoption across multiple industries.
The implementation of Rapid Microbial Methods represents a paradigm shift in microbiological quality control, offering significant advantages over traditional culture-based methods in speed, sensitivity, and automation potential. While barriers related to cost, validation complexity, and workforce readiness have historically slowed adoption, current trends indicate accelerating acceptance driven by technological advancements, compelling operational benefits, and evolving regulatory frameworks.
The comparative experimental data presented in this analysis demonstrates that modern RMM platforms like the Sievers Soleil Rapid Bioburden Analyzer can deliver equivalent or superior performance to traditional methods while dramatically reducing time-to-results from days to minutes [14]. As AI integration advances, molecular technologies evolve, and portable platforms become more sophisticated, the adoption of RMMs is expected to accelerate further across pharmaceutical, biopharmaceutical, food safety, and environmental monitoring applications.
For researchers, scientists, and drug development professionals, understanding these trends and implementation considerations is crucial for making informed decisions about RMM adoption. The comprehensive validation protocols, performance comparison data, and emerging technology insights provided in this article offer a foundation for evaluating these transformative technologies and leveraging their potential to enhance product safety, improve operational efficiency, and advance public health protection.
Rapid Method Technologies in Action: PCR, NGS, ATP, and AI-Driven Platforms
The field of microbiological testing is undergoing a significant transformation, moving from traditional culture-based methods toward rapid molecular techniques. Traditional methods, while well-established and reliable, often require 48 to 72 hours or longer for results due to the need for microbial incubation, creating delays in critical decision-making for clinical diagnostics and product safety testing [1]. In this context, Polymerase Chain Reaction (PCR) and Next-Generation Sequencing (NGS) have emerged as powerful, rapid methods for pathogen identification. These techniques offer not only speed but also enhanced sensitivity and specificity, enabling the detection of low-abundance pathogens and complex genetic characterization that were previously challenging or impossible [22]. This guide provides an objective comparison of PCR and NGS performance, supported by experimental data, to help researchers select the appropriate method for their pathogen identification needs.
Fundamental Principles
Real-Time PCR (qPCR) is a targeted molecular technique that amplifies and detects specific DNA sequences simultaneously. It relies on the principle of monitoring fluorescence at each amplification cycle, allowing for both detection and quantification of the target pathogen. The cycle threshold (Ct) value, which indicates the amplification cycle at which fluorescence crosses a background threshold, is inversely proportional to the amount of target nucleic acid in the sample [23] [24]. This method is exceptionally sensitive, capable of detecting as few as 1.3 copies of a target gene in a 50-μl reaction [24].
Next-Generation Sequencing (NGS) represents a fundamentally different approach, enabling massively parallel sequencing of millions of DNA fragments simultaneously. Unlike PCR's targeted analysis, NGS can perform unbiased sequencing of all nucleic acids in a sample, allowing for comprehensive pathogen detection without prior knowledge of potential contaminants [25] [26]. This makes it particularly valuable for discovering novel pathogens or identifying complex microbial communities. Two primary NGS approaches are used in transcriptome analysis: RNA-Seq for detecting all cellular RNA types, and targeted transcriptome sequencing for focusing on known mRNA transcripts [23].
Experimental Workflows
The typical workflows for PCR and NGS involve distinct processes from sample preparation to data analysis, each with specific requirements and outputs.
Figure 1: Comparative Workflows of PCR and NGS for Pathogen Identification
Performance Comparison: Experimental Data and Case Studies
Detection Sensitivity and Limit of Detection (LoD)
Sensitivity varies significantly between PCR and NGS depending on the specific application, target pathogen, and experimental conditions.
Table 1: Comparison of Detection Sensitivity Between PCR and NGS
| Pathogen/Application | PCR Performance | NGS Performance | Experimental Context |
|---|---|---|---|
| Helicobacter pylori | Detected in 16/40 samples (40.0%) [27] | Detected in 14/40 samples (35.0%) [27] | Pediatric gastric biopsies; both methods showed similar detection rates |
| EGFR mutations (L858R) | Detected at 3% Variant Allele Fraction (VAF) [28] | Detected at 3% VAF with read depth of 2672 [28] | Non-small cell lung cancer (NSCLC) specimens |
| EGFR exon 19 deletion | Detected at 1% VAF (deltaCt 7.62) [28] | Failed detection at 0.8% VAF (below LoD) [28] | NSCLC specimens; PCR showed superior sensitivity for this specific mutation |
| EGFR T790M mutation | Limited detection (high LoD: 17.5% VAF); late Ct curves unreliable [28] | Detected at 2-5% VAF [28] | NSCLC specimens; NGS showed superior sensitivity for this mutation |
| Mycoplasma contamination | Limited by non-specific amplification with E. rhusiopathiae [25] | 100-fold lower detection limits depending on species [25] | Veterinary vaccine quality control |
The data reveals that sensitivity is context-dependent. In some scenarios, such as detecting EGFR exon 19 deletions in NSCLC, PCR demonstrates superior sensitivity for low-abundance targets (1% VAF) compared to NGS, which failed to detect the variant at 0.8% VAF [28]. Conversely, for other targets like the EGFR T790M mutation, NGS outperforms PCR by detecting variants at 2-5% VAF that were missed by the PCR assay [28]. This complementary sensitivity profile highlights the importance of understanding the specific analytical requirements for each application.
Multiplexing Capability and Target Range
The fundamental difference between PCR and NGS lies in their scope of detection. PCR is ideal for targeted analysis of known pathogens, while NGS excels at comprehensive profiling of diverse microbial communities.
Table 2: Multiplexing Capability and Applications
| Feature | PCR | NGS |
|---|---|---|
| Multiplexing Capacity | Limited (typically < 10 targets per reaction) | High (thousands to millions of sequences simultaneously) |
| Discovery Potential | Limited to known targets with predefined primers | High capability for novel pathogen discovery |
| Ideal Application | Routine screening of specific pathogens | Metagenomic studies, outbreak investigation, unknown pathogen identification |
| Experimental Evidence | EGFR therascreen test detects 21 predefined mutations [28] | Identified complex viral replication/recombination dynamics in HBV [29] |
| Data Output | Quantitative (Ct values) for specific targets | Sequencing reads, variant identification, phylogenetic relationships |
In veterinary vaccine testing, NGS demonstrated particular utility when PCR failed due to non-specific amplification. When testing combination vaccines containing Erysipelothrix rhusiopathiae, PCR produced non-specific bands that complicated interpretation, while NGS accurately differentiated between Mycoplasma species and the vaccine component through a reference-mapping method that filtered non-specific reads [25]. This demonstrates NGS's advantage in complex samples where multiple similar sequences may be present.
Turnaround Time and Operational Considerations
Table 3: Operational Characteristics and Workflow Requirements
| Parameter | PCR | NGS |
|---|---|---|
| Hands-on Time | Minimal after setup | Extensive for library preparation and data analysis |
| Total Turnaround Time | 1-2 days for most applications [23] | 1-3 days for entire workflow [23] [29] |
| Throughput | Medium (typically 20 samples and 10 targets in 1-2 days) [23] | High (96 samples sequenced in 14 hours) [29] |
| Technical Expertise | Standard molecular biology skills | Requires bioinformatics expertise for data analysis |
| Cost per Sample | Lower for small target numbers | Higher, but more cost-effective for multiple targets |
| Automation Potential | High for established assays | Medium, with increasing automation options |
While NGS is often perceived as slower, technological advances have significantly reduced sequencing times. For example, a study on hepatitis B virus (HBV) utilizing nanopore sequencing generated whole-genome data for 96 samples within 14 hours, nearly twice as fast as legacy short-read technologies [29]. The same study highlighted that nanopore sequencing required fewer reagents and enabled real-time data analysis, streamlining the workflow further.
Detailed Experimental Protocols
PCR Protocol for Pathogen Detection (Based on H. pylori Detection)
The following protocol is adapted from studies comparing real-time PCR and NGS for Helicobacter pylori detection in pediatric gastric biopsies [27]:
Sample Collection and DNA Extraction:
- Collect gastric biopsy specimens and preserve in appropriate transport medium.
- Subject tissue samples to mechanical lysis for 1 minute using a manual homogenizer.
- Digest samples in 200 μL of trypsin solution (5 mg/mL) at 37°C for 30 minutes to increase DNA isolation efficiency.
- Perform DNA extraction using a commercial pathogen DNA isolation kit according to manufacturer's instructions.
- Store isolated DNA at -20°C until analysis.
Real-Time PCR Setup:
- Utilize an IVD-certified real-time PCR kit specific for the target pathogen.
- Prepare reaction mix according to manufacturer's specifications, typically containing:
- Master mix (including DNA polymerase, dNTPs, buffer)
- Target-specific primers and probes
- Template DNA (2 μL per reaction)
- Perform amplification using standard cycling conditions:
- Initial denaturation: 95°C for 10 minutes
- 40-45 cycles of:
- Denaturation: 95°C for 15 seconds
- Annealing/Extension: 60°C for 1 minute
- Analyze results based on Ct values, with samples below a predetermined threshold considered positive.
Quality Control:
- Include positive and negative controls in each run.
- Verify assay performance using internal controls when available.
Targeted NGS Protocol for Pathogen Identification (Based on HBV Sequencing)
This protocol is adapted from a study utilizing nanopore sequencing for hepatitis B virus characterization [29]:
Library Preparation:
- Perform multiplexed tiled PCR to amplify the entire pathogen genome.
- Utilize primers designed to generate overlapping amplicons covering the complete genomic sequence.
- Purify PCR products using magnetic bead-based clean-up systems.
- Quantify DNA using fluorometric methods and normalize concentrations.
Sequencing:
- Prepare sequencing libraries using a ligation sequencing kit according to manufacturer's instructions.
- Load the library onto a nanopore sequencing device (such as GridION).
- Initiate sequencing run with real-time base calling enabled.
- Continue sequencing until desired coverage is achieved (approximately 2,300x coverage over >80% of genome).
Data Analysis:
- Perform base calling and demultiplexing using native instrument software.
- Conduct reference-based alignment to map reads to the target pathogen genome.
- Identify variants and reconstruct complete genomes.
- Perform genotyping and detect drug resistance mutations through comparison with established databases.
Research Reagent Solutions
Table 4: Essential Research Reagents for PCR and NGS Workflows
| Reagent/Kit | Function | Application Examples |
|---|---|---|
| Streck Cell Free DNA BCT Tubes | Stabilizes blood samples for cell-free DNA analysis | ctDNA detection in rectal cancer studies [30] |
| Ion AmpliSeq Cancer Hotspot Panel v2 | Targeted NGS library preparation for cancer-associated mutations | Identification of somatic alterations in rectal tumor specimens [30] |
| TaqMan Gene Expression Assays | Target-specific probes for real-time PCR detection | Verification of NGS results; targeted gene expression analysis [23] |
| GeneProof PathogenFree DNA Isolation Kit | Nucleic acid extraction from clinical specimens | DNA isolation from gastric biopsies for H. pylori detection [27] |
| Nanopore Sequencing Kits (Ligation Sequencing) | Preparation of libraries for long-read sequencing | Whole-genome sequencing of HBV viral amplicons [29] |
PCR and NGS offer complementary strengths for pathogen identification, and the choice between them should be guided by specific research requirements:
Choose PCR when: The target pathogens are known, high sensitivity for specific mutations is required, rapid turnaround time is critical, and budget constraints exist. PCR is particularly effective for detecting low-abundance variants like EGFR exon 19 deletions at 1% VAF [28] and for routine diagnostic applications where predefined targets are monitored.
Choose NGS when: Comprehensive pathogen discovery is needed, multiple unknown pathogens may be present, complex genetic characterization is required, or when investigating cases with ambiguous results from targeted methods. NGS demonstrates superior capability for identifying recombinant viral strains [29], detecting contamination in complex samples [25], and providing complete genomic information.
The most effective pathogen identification strategy often involves leveraging both technologies synergistically - using PCR for rapid, sensitive detection of known targets and NGS for comprehensive analysis and discovery of novel or unexpected pathogens. As one study noted, "NGS could complement PCR in diagnosing difficult or ambiguous cases, enabling the detection of multiple pathogens simultaneously" [27].
Table of Contents
- Introduction: The Shift to Rapid Methods
- Principles of Detection
- Experimental Protocols in Practice
- Performance Comparison: Data-Driven Analysis
- Advantages, Limitations, and Suitability
- Conclusion: Selecting the Right Tool
The accurate assessment of cell viability is a critical quality attribute in biomedical research, drug development, and cellular therapy manufacturing [31]. For decades, the field relied on traditional culture-based methods, such as the colony-forming unit (CFU) count and the chromium-51 (51Cr) release assay. While these are considered the historical "gold standard," they are hampered by being time-consuming, labor-intensive, and slow to provide results—often taking from several days to two weeks [13] [7]. The 51Cr assay also involves radioactive materials, posing health and disposal challenges [32].
This landscape is rapidly transforming with the adoption of Rapid Microbiological Methods (RMMs), which offer faster, more sensitive, and higher-throughput alternatives [7]. Among the most prominent RMMs for viability assessment are ATP bioluminescence and flow cytometry. These methods align with the needs of modern laboratories for timely results, essential for ensuring the safety and efficacy of products like cell therapies, where patients may need immediate treatment [33]. This guide provides an objective comparison of ATP bioluminescence and flow cytometry, equipping researchers to select the optimal method for their specific application.
Principles of Detection
ATP bioluminescence and flow cytometry operate on distinct biochemical and physical principles to determine cell viability.
ATP Bioluminescence: This method leverages the fact that adenosine triphosphate (ATP) is a universal energy currency present in all metabolically active cells. The assay utilizes the firefly luciferase enzyme, which catalyzes a reaction between luciferin and oxygen, requiring ATP as a cofactor. The amount of light produced (bioluminescence) is directly proportional to the concentration of ATP, which in turn is directly proportional to the number of viable cells present [34] [35] [36]. A dying or dead cell rapidly loses its ATP content and ceases to produce a signal.
Flow Cytometry: This technique assesses viability by exploiting the integrity of the cell membrane. Viable cells possess intact membranes that exclude certain dyes. Nucleic acid-binding dyes like propidium iodide (PI) and 7-aminoactinomycin D (7-AAD) are impermeant to live cells. However, when a cell dies, its membrane becomes compromised, allowing these dyes to enter, bind to DNA/RNA, and fluoresce brightly [31]. By passing cells single file through a laser beam and detecting this fluorescence, a flow cytometer can precisely quantify the proportion of non-viable cells in a population. Furthermore, flow cytometry can simultaneously analyze cell surface or intracellular markers, enabling viability assessment within specific cell subpopulations [31].
The following diagram illustrates the core signaling pathways and workflows for these two detection methods.
Experimental Protocols in Practice
ATP Bioluminescence Assay Protocol
The ATP assay is a homogeneous, endpoint assay known for its simplicity and speed [34] [37].
- Cell Seeding and Treatment: Plate cells in a multi-well plate (e.g., 96-well) and apply the experimental treatment.
- Equilibration: Allow the assay reagent, such as CellTiter-Glo, to equilibrate to room temperature.
- Reagent Addition: Add a volume of the single-step, homogeneous reagent equal to the volume of cell culture medium present in each well.
- Mixing and Lysis: Mix the contents on an orbital shaker for 2-5 minutes to induce cell lysis and stabilize the luminescent signal.
- Signal Measurement: Transfer the lysate to an opaque plate and measure the luminescence using a plate-reading luminometer. The signal, expressed in Relative Light Units (RLU), is directly proportional to the amount of ATP present and, thus, the number of viable cells.
Flow Cytometry Viability Staining Protocol
This protocol is more complex and requires single-cell suspensions [31].
- Cell Harvesting: Harvest cells and wash them with a cold buffer like phosphate-buffered saline (PBS).
- Staining Preparation: Resuspend the cell pellet in a staining buffer. For direct staining with a dye like 7-AAD or PI, add the appropriate volume of dye to the cell suspension.
- Incubation: Incubate the mixture for 5-20 minutes at room temperature in the dark to prevent dye degradation and photobleaching.
- Analysis: Without a wash step (to avoid losing dead cells), acquire the samples immediately on a flow cytometer. For assays combining viability staining with cell surface marker detection, stain for surface antigens first with fluorochrome-labeled antibodies for 20 minutes at 4°C, wash the cells, then perform a viability stain with 7-AAD before acquisition [31].
- Gating Strategy: During analysis, create a plot of forward scatter (FSC-A) versus side scatter (SSC-A) to gate on the cell population of interest. Then, on a histogram or dot plot displaying fluorescence for 7-AAD or PI, gate to distinguish the negative (viable) and positive (non-viable) populations.
Performance Comparison: Data-Driven Analysis
The following tables summarize key performance metrics and experimental data for ATP bioluminescence and flow cytometry, drawing from direct comparisons and validation studies.
Table 1: Direct Method Comparison Based on Technical and Operational Factors
| Parameter | ATP Bioluminescence | Flow Cytometry |
|---|---|---|
| Detection Principle | Metabolic activity (ATP content) | Membrane integrity (Dye exclusion) |
| Measured Signal | Luminescence (RLU) | Fluorescence |
| Assay Format | Homogeneous, bulk population | Single-cell analysis |
| Throughput | Very High (96/384-well plates) | Moderate to High |
| Speed of Analysis | Very Fast (minutes after reagent addition) | Fast (minutes to acquire, longer for complex panels) |
| Multiplexing Capability | Low (typically standalone) | High (can combine with immunophenotyping) |
| Sample Requirement | Bulk lysate | Single-cell suspension |
| Key Instrument | Luminometer | Flow Cytometer |
Table 2: Experimental Performance Data from Comparative Studies
| Study Context & Citation | ATP Bioluminescence Performance | Flow Cytometry Performance |
|---|---|---|
| Glioma Drug Screens [34] | Superior for viability; smaller standard deviations vs. NADH-based assays. Robust for most treatments, but caution with metabolism-interfering drugs. | Not the primary focus, but apoptosis/cytotoxicity assays (often flow-based) did not unequivocally detect responses in this context. |
| Cellular Therapy Products [31] | - | Highly reliable for fresh and cryopreserved products (PBMCs, CAR-T cells) using 7-AAD/PI. Enabled viability tracking in specific immune cell subsets. |
| Cytotoxicity Measurement [32] | BLI-based ATP assay outperformed 51Cr: superior robustness, signal-to-noise ratio, and faster kinetics. | Flow cytometric methods using CFSE/PI showed similar or higher sensitivity than 51Cr. |
| BCG Vaccine Potency [38] | Strong linear correlation with CFU (r²=0.9874 for BCG Bulk). Validated as a rapid, sensitive alternative to slow colony counts. | - |
Advantages, Limitations, and Suitability
The choice between ATP bioluminescence and flow cytometry is dictated by the specific research question, as each method has distinct strengths and weaknesses.
ATP Bioluminescence is the preferred tool for high-throughput screening applications where the primary goal is to rapidly assess the viability or proliferation of a homogeneous cell population in response to many compounds [34] [36]. Its key strengths are speed, sensitivity, ease of use, and robust, reproducible data. Its main limitation is that it provides a bulk population measurement and cannot distinguish viability within specific cell subsets in a mixed culture. Furthermore, treatments that directly affect cell metabolism (e.g., mTOR inhibitors like rapamycin) can confound results, as the readout is ATP levels [34].
Flow Cytometry is the unequivocal choice when multiparameter analysis is required [31]. Its principal advantage is the ability to simultaneously measure viability and identify specific cell types via antibody staining (e.g., assessing T-cell viability within total PBMCs). It provides deep, single-cell insights. The drawbacks include higher operational complexity, the need for specialized and costly instrumentation, and a generally lower throughput compared to plate-based luminescence assays. The requirement for a single-cell suspension can also be a limiting factor for some sample types.
Both ATP bioluminescence and flow cytometry are powerful, rapid methods that have largely superseded traditional, slower techniques for viability assessment. The decision between them is not about which is universally better, but which is fit-for-purpose.
- For high-throughput drug screens or rapid potency assays where the end goal is a simple, quantitative measure of total viable cells, ATP bioluminescence offers an efficient and robust solution.
- For characterizing complex cellular products (like CAR-T cells or PBMCs), where understanding the health of specific subpopulations is critical, flow cytometry provides indispensable, multi-dimensional data.
Researchers must weigh factors such as throughput, required information depth, sample type, and available resources. By understanding the principles, protocols, and performance characteristics detailed in this guide, scientists can make an informed choice that enhances the efficiency, accuracy, and depth of their contamination detection and viability research.
Research Reagent Solutions
Table 3: Essential Materials and Reagents for Viability Detection
| Item | Function / Application | Specific Examples |
|---|---|---|
| Homogeneous Viability Assay Kits | One-step reagent for ATP-based luminescent detection in plate formats. | CellTiter-Glo Luminescent Cell Viability Assay [34] |
| Viability Staining Dyes | Nucleic acid dyes for flow cytometry; excluded by viable cells. | Propidium Iodide (PI), 7-Aminoactinomycin D (7-AAD) [31] |
| Fluorochrome-conjugated Antibodies | For immunophenotyping to identify cell subtypes during flow analysis. | Anti-CD3, Anti-CD45, Anti-CD19 [31] |
| Cell Culture Media | Supports growth of specific cell types, including stem-like cells. | Serum-free neural stem cell media [34] |
| Extracellular Matrix Coating | Provides surface for adherent cell growth in viability assays. | Growth factor-reduced ECM (e.g., from BD Biosciences) [34] |
Mass Spectrometry and Biosensors for High-Specificity Analysis
In the field of pharmaceutical quality control and contamination research, the limitations of traditional microbiological methods—primarily their extended time-to-results—have driven the adoption of rapid, high-specificity technologies [1] [39]. Traditional methods, like the standard plate count, often require 48 to 72 hours for initial results, with sterility testing demanding up to 14 days of incubation [8] [22]. This delay is ill-suited for modern manufacturing, where quicker feedback is essential for product safety and supply chain efficiency [39].
Mass spectrometry and biosensors represent a paradigm shift, offering sensitive, precise, and reproducible results in a significantly shorter timeframe [22]. These technologies enable researchers to move from merely detecting contamination to precisely identifying and quantifying specific contaminants, supporting more robust quality control and faster decision-making [40] [39]. This guide provides a detailed comparison of these advanced platforms, focusing on their application in high-specificity analysis for pharmaceutical contamination detection.
The following table summarizes the core characteristics of the main rapid methods discussed in this guide.
Table 1: Comparison of High-Specificity Analytical Platforms
| Technology Platform | Key Principle | Typical Time to Result | Key Advantages | Primary Applications in Contamination Detection |
|---|---|---|---|---|
| Mass Spectrometry (e.g., MALDI-TOF) | Ionizes sample molecules and separates them based on mass-to-charge ratio [40] | Minutes to hours for identification [7] | High sensitivity and resolution; can identify molecules up to 100,000 u [41]; excellent for characterizing complex biological interactions [40] | Rapid microbial identification [7], characterization of biomolecules (proteins, sugars) [40], biosensor surface characterization [41] |
| SERS Biosensors | Enhances Raman scattering signal of target molecules using plasmonic nanostructures [42] | Minutes to hours (often requires incubation) [42] | Extremely high sensitivity (single-molecule potential); capable of multiplexing; uses intrinsic vibrational modes of biomarkers [42] | Detection of specific biomarkers (e.g., α-fetoprotein for cancer) [42], pathogen detection, mycotoxin analysis |
| Electrochemical Biosensors | Measures electrical changes (current, impedance) due to biorecognition event on electrode surface [42] | Minutes to hours [42] | High potential for portability and miniaturization; suitable for wearable devices; enzyme-free designs available [42] | Glucose monitoring, detection of environmental pollutants (pesticides, heavy metals) in samples [42] |
| PCR & Molecular Methods | Amplifies specific DNA/RNA sequences for detection [1] [8] | Several hours [8] | High specificity and sensitivity; can detect low levels of specific pathogens [1] [8] | Detection of specific pathogens, viral contamination, and viable but non-culturable (VBNC) organisms [8] |
A critical difference between mass spectrometry and biosensors lies in their operational approach. Biosensors are typically designed for specificity first, using immobilized biological elements (antibodies, aptamers, enzymes) to capture a single target or a predefined set of targets [40] [42]. Mass spectrometry, particularly when coupled with separation techniques, is a discovery-oriented tool that can profile all ionizable molecules in a sample without prior specification, making it powerful for identifying unknown contaminants [40] [43].
Mass Spectrometry-Based Analysis
Key Experimental Protocols
Mass spectrometry-based biosensing encompasses several sophisticated techniques for biomolecular analysis [40]:
Array-Based Sensing with MALDI-MS: This protocol involves immobilizing probe molecules (e.g., peptides, antibodies) on a functionalized surface or biochip to capture target analytes from a complex sample. After washing, a matrix is applied to the chip, which is then analyzed by Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry (MALDI-MS). The mass spectrometer detects the captured targets or mass-tagged reporter molecules, enabling multiplexed and highly sensitive detection [40]. A specific application is the SAMDI-MS technique, which uses self-assembled monolayers (SAMs) on gold chips to study enzyme activity and screen inhibitors with high throughput [40].
Probe-Based Micro-Sampling for In-Situ Analysis: For in-situ or in-vivo analysis, specialized probes (e.g., sharp metal needles, coated fibers) are used to collect trace samples directly from surfaces or tissues. The collected material is then ionized directly from the probe tip using ambient ionization techniques like Desorption Electrospray Ionization (DESI) or paper spray ionization, and analyzed by a mass spectrometer. This method minimizes sample preparation and allows for spatial profiling of biomolecules [40].
Coupling with Other Biosensors (e.g., SPR-MS): This method couples the sensitive affinity capture of a biosensor like Surface Plasmon Resonance (SPR) with the identification power of mass spectrometry. The target analyte is first captured from the sample by the biosensor chip. The specific area where binding occurred is then treated with a matrix and analyzed by MALDI-MS, confirming the identity of the captured molecule and validating the biosensor's response [40].
The workflow for a typical MALDI-MS based biosensing experiment is visualized below.
Essential Research Reagent Solutions
Table 2: Key Reagents for Mass Spectrometry-Based Biosensing
| Reagent / Material | Function in the Experimental Protocol |
|---|---|
| Functionalized Biochips (e.g., SAM-Au chips, ITO glass) | Provides a solid substrate for immobilizing probe molecules (antibodies, peptides) in an ordered array for specific target capture [40]. |
| Mass Tags (e.g., unique metal nanoparticles, stable isotopes) | Serve as reporter molecules that can be detected by MS, enabling signal amplification and multiplexed detection of multiple targets simultaneously [40]. |
| Ionization Matrix (e.g., CHCA, Sinapinic Acid) | A chemical compound that absorbs laser energy and facilitates the soft desorption and ionization of the target analyte for MALDI-MS analysis [40]. |
| Self-Assembled Monolayer (SAM) Reagents | Alkanethiols that form ordered monolayers on gold surfaces, enabling precise engineering of surface properties for biomolecule attachment [40]. |
Biosensor-Based Analysis
Key Experimental Protocols
Biosensors integrate a biological recognition element with a transducer that converts the binding event into a measurable signal [40]. Key experimental protocols include:
Surface-Enhanced Raman Scattering (SERS) Immunoassay: This protocol begins with the synthesis and optimization of plasmonic nanoparticles (e.g., gold-silver nanostars). These nanostars are then functionalized with a Raman reporter and a capture antibody specific to the target (e.g., α-fetoprotein). The sample is incubated with the functionalized nanoparticles, allowing the target antigen to bind. After washing, the SERS signal is measured directly from the intrinsic vibrations of the captured antigen or the reporter molecule, providing highly sensitive and specific detection [42].
Electrochemical Aptasensor Fabrication: This involves modifying an electrode surface with a conductive polymer or nanostructured material (e.g., porous gold, polyaniline). An aptamer—a single-stranded DNA or RNA molecule that binds a specific target—is then immobilized on this electrode. Upon exposure to the sample, the target molecule binds to the aptamer, causing a conformational change that alters the electrical properties (e.g., current, impedance) at the electrode surface, which is measured quantitatively [42].
The workflow for a SERS-based biosensing experiment is detailed below.
Essential Research Reagent Solutions
Table 3: Key Reagents for Biosensor-Based Analysis
| Reagent / Material | Function in the Experimental Protocol |
|---|---|
| Plasmonic Nanoparticles (e.g., Au-Ag Nanostars) | Enhance the Raman scattering signal by several orders of magnitude, forming the core of highly sensitive SERS biosensors [42]. |
| Aptamers or Monoclonal Antibodies | Act as the biological recognition element that provides high specificity by binding exclusively to the target analyte (e.g., a pathogen or biomarker) [42]. |
| Electrode Modification Materials (e.g., Polydopamine, Polyaniline) | Used to coat and functionalize electrode surfaces, improving biocompatibility, stability, and electrical conductivity for electrochemical sensors [42]. |
| Coupling Agents (e.g., EDC, NHS) | Crosslinking chemicals used to covalently immobilize antibodies or other biomolecules onto sensor surfaces or nanoparticles [42]. |
The transition from traditional, growth-based methods to rapid, high-specificity technologies is fundamentally enhancing contamination detection research. Mass spectrometry and biosensors are not mutually exclusive but are often complementary. The choice between them depends on the research goal: biosensors are ideal for routine, high-throughput screening of known analytes, while mass spectrometry excels in discovery, identification of unknowns, and multiplexed profiling. As these technologies continue to evolve, their integration into pharmaceutical quality control promises faster release times, improved patient safety, and a deeper understanding of contamination events [39] [8].
AI and Automation in Agar Plate Reading and Data Interpretation
In the field of microbiological contamination detection, a significant transformation is underway, moving from traditional, labor-intensive methods toward automated, data-driven approaches. For decades, traditional growth-based methods have served as the gold standard for microbial detection, relying on visual inspection of culture plates after prolonged incubation periods [16] [44]. While these methods provide valuable data, they introduce substantial limitations including operator subjectivity, lengthy time-to-results (often 3-14 days), and inherent variability in interpretation [8] [44]. The emergence of rapid microbiological methods (RMMs) represents a fundamental shift in this landscape, with artificial intelligence (AI) and automation now enabling unprecedented levels of efficiency, objectivity, and data richness in agar plate reading and interpretation [45] [46].
This evolution aligns with broader trends in pharmaceutical and clinical microbiology, where laboratories are increasingly adopting technologies that enhance throughput while reducing human error [47]. AI-powered imaging systems can now process hundreds of plates per hour, automatically distinguishing between contaminated and sterile samples with consistent accuracy [46]. This transition from subjective human interpretation to algorithm-driven analysis represents not merely an incremental improvement but a fundamental reimagining of microbiological quality control processes, particularly crucial for sterility testing in pharmaceutical manufacturing and clinical diagnostics [46] [44].
Comparative Analysis: Traditional vs. Automated Methods
Performance Metrics and Experimental Data
The comparison between traditional and AI-automated plate reading methods reveals significant differences across multiple performance dimensions. The following table summarizes key quantitative comparisons based on experimental data and implementation studies:
Table 1: Performance Comparison of Traditional vs. Automated Plate Reading Methods
| Performance Metric | Traditional Manual Reading | AI-Automated Systems | Experimental Support |
|---|---|---|---|
| Throughput Capacity | ~20-40 plates per technologist hour [46] | ~200 plates per hour [46] | Clever Culture Systems APAS Independence implementation data |
| Time to Result | 3-14 days for growth-based methods [8] [44] | Hours for initial screening [8] | Pharmaceutical validation studies |
| Operator Subjectivity | High (inter-operator variability) [44] | Minimal (algorithm consistency) [46] [8] | Comparative studies on colony enumeration |
| Error Rate | Variable (false positives/negatives reported) [44] | Significantly reduced [46] | Environmental monitoring validation |
| Documentation | Manual record-keeping | Automated digital archiving [46] | Regulatory compliance assessments |
| Continuous Operation | Limited by staffing | 24/7 operation possible [47] | Laboratory workflow analyses |
Detection Capabilities and Technological Advancements
Beyond throughput and efficiency metrics, automated systems demonstrate enhanced technical capabilities for microbial detection:
Table 2: Detection Capabilities of Microbial Methods
| Detection Aspect | Traditional Methods | Rapid/AI Methods | Technical Basis |
|---|---|---|---|
| Detection Principle | Visual colony observation [8] | AI image analysis, markers (ATP, DNA) [8] | Technology specifications |
| Viable but Non-Culturable Detection | Limited [8] | Enhanced (via alternative markers) [8] | Comparative detection studies |
| Quantification Approach | Colony-forming units (CFUs) [44] | Relative light units, digital counts [8] | Method validation data |
| Multiplexing Capability | Limited | Moderate to high [48] | System specifications |
| Data Integration | Manual entry | Direct LIMS integration [8] | Workflow studies |
Methodologies and Experimental Protocols
Traditional Culture-Based Plate Reading Protocol
The conventional approach to agar plate reading follows standardized methodology with minimal technological intervention:
Materials and Reagents:
- Prepared culture media (TSA, SDA, or specialized formulations)
- Incubators (controlled temperature 30-35°C for bacteria, 20-25°C for fungi)
- Biological safety cabinet
- Colony counter (manual or semi-automated)
- Recording materials (laboratory notebooks, spreadsheets)
Experimental Procedure:
- Inoculation and Incubation: Samples are streaked or spread onto agar surfaces under aseptic conditions and incubated for specified durations (typically 3-5 days for bacteria, 5-7 days for fungi) [44].
- Visual Inspection: Technologists examine plates daily for growth characteristics, recording colony morphology, size, color, and enumeration.
- Colony Counting: Manual counting of CFUs using colony counters, with subjective determination of colony differentiation.
- Interpretation and Reporting: Results are transcribed manually into laboratory records, with classification based on established criteria.
Limitations Documented in Experimental Studies:
- Subjectivity: Inter-technologist variation in colony identification and counting [44].
- Time Delays: Extended incubation requirements delay result availability and decision-making [8].
- Documentation Challenges: Manual transcription introduces potential for reporting errors [46].
AI-Automated Plate Reading Protocol
The automated workflow incorporates sophisticated imaging and machine learning algorithms:
Materials and Reagents:
- Prepared culture media (compatible with imaging systems)
- Automated plate imaging system (e.g., Clever Culture Systems APAS Independence)
- Computer workstation with AI analysis software
- Laboratory Information Management System (LIMS)
Experimental Procedure:
- Plate Loading: Plates are automatically fed into the imaging system following incubation [46] [49].
- High-Resolution Imaging: System captures multiple images under different lighting conditions to enhance detection sensitivity [49].
- AI Analysis: Machine learning algorithms process images to:
- Result Verification: System flags uncertain results for human technologist review.
- Automated Reporting: Results are digitally transmitted to LIMS with complete image documentation [46].
Validation Studies: Pharmaceutical implementations have demonstrated that automated systems can process 200 plates per hour while maintaining compliance with regulatory requirements for sterility testing and environmental monitoring [46].
The Scientist's Toolkit: Essential Research Reagents and Solutions
Implementation of AI and automation in agar plate reading requires specific technological solutions and reagents:
Table 3: Research Reagent Solutions for Automated Plate Reading
| Item | Function | Application Notes |
|---|---|---|
| APAS PharmaQC Software | AI-based image analysis for pharmaceutical QC | Specifically validated for pharma environments; FDA-cleared [46] |
| Specialized Culture Media | Optimized for automated imaging | Formulations with minimal interference for image analysis |
| Validation Strains | System performance qualification | Reference microorganisms for algorithm training |
| Data Integration Modules | LIMS connectivity | Ensures regulatory compliance and data integrity [46] |
| Calibration Standards | Imaging system calibration | Maintains measurement accuracy over time |
| Machine Learning Algorithms | Colony recognition and classification | Trained on diverse microbial morphologies [49] |
Signaling Pathways and Workflow Visualization
The integration of AI and automation creates sophisticated workflows that combine physical processes with digital analysis:
Automated Plate Reading Workflow
This integrated process demonstrates how modern systems combine physical microbiology practices with digital analysis, creating a seamless workflow that maximizes efficiency while maintaining necessary human oversight for complex cases.
Implementation Case Studies and Validation Data
Pharmaceutical Industry Adoption
Major pharmaceutical companies have begun implementing AI-based automated reading systems with documented results:
AstraZeneca Implementation:
- Challenge: Need for objective, efficient quality control in pharmaceutical manufacturing [46].
- Solution: Development and implementation of APAS Pharma analysis module specifically for pharmaceutical quality control [46].
- Results: Successful testing across environmental monitoring plates, sterility test plates, and microbial limits testing [46].
- Outcome: System adoption in August 2024 following validation studies [46].
Bristol Myers Squibb Implementation:
- Timeline: Technology evaluation throughout 2024, with adoption in October 2024 [46].
- Application: Focus on 90mm culture plates and development of 55mm contact plates [46].
- Significance: Second major pharmaceutical company adoption within months, indicating growing industry acceptance [46].
Performance Validation Metrics
Independent studies and validation reports demonstrate specific performance advantages:
Efficiency Metrics:
- Throughput: Automated systems process approximately 200 plates per hour compared to 20-40 plates with manual reading [46].
- Labor Reduction: Automatic reporting of negative plates removes approximately 70-80% of plates from manual workflow [49].
- Time Savings: Reduction in result reporting time from days to hours for negative samples [8].
Quality Metrics:
- Consistency: Elimination of inter-operator variability in colony counting and interpretation [46].
- Documentation: Automated digital archiving of all plate images for regulatory compliance and data integrity [46].
- Sensitivity: Enhanced detection of small or faint colonies through optimized imaging parameters [49].
Future Directions and Developing Trends
The field of automated agar plate reading continues to evolve with several emerging trends:
Integration with Broader Laboratory Ecosystems: Automated plate reading systems are increasingly becoming integration points within larger laboratory workflows, connecting with environmental monitoring systems, manufacturing execution systems, and quality management platforms [47]. This integration enables comprehensive contamination control strategies rather than isolated testing procedures.
Regulatory Evolution: As automated systems demonstrate consistent performance, regulatory bodies are developing appropriate frameworks for their validation and implementation [46]. The FDA clearance of the APAS system establishes an important precedent for future innovations [46].
Advanced Analytics and Predictive Applications: The accumulation of digital plate reading data creates opportunities for advanced analytics, including trend analysis, predictive contamination modeling, and root cause investigation support [48]. Machine learning algorithms continue to improve through expanded training datasets and advanced neural network architectures.
The integration of AI and automation into agar plate reading represents a fundamental advancement in microbiological detection methodology. Quantitative comparisons demonstrate clear advantages in throughput, consistency, and efficiency compared to traditional manual methods. The documented implementations by leading pharmaceutical companies provide compelling evidence of the technology's maturity and regulatory acceptance.
While traditional growth-based methods remain the compendial standard and retain value for certain applications, the demonstrated benefits of automated systems position them as the future foundation for microbiological quality control in pharmaceutical manufacturing and clinical diagnostics. As these technologies continue to evolve and accumulate validation data, their adoption is expected to accelerate, ultimately transforming how the scientific community approaches microbial detection and contamination control.
In the demanding fields of pharmaceutical manufacturing and drug development, ensuring microbiological control is paramount for patient safety and product quality. For decades, this reliance has been placed on traditional culture-based methods, which, while well-established, require extended incubation periods of up to 14 days for sterility tests and several days for bioburden results [50] [51]. This delay forces companies to release products at risk or halt production, creating significant operational inefficiencies. The limitations of traditional methods have catalyzed the development and adoption of Rapid Microbiological Methods (RMMs), which leverage advanced technologies to provide faster, more sensitive, and often automated detection of microorganisms [14] [7]. This guide provides an objective comparison of these methodologies, supported by experimental data, to inform researchers and scientists in their pursuit of optimal contamination control strategies.
Performance Comparison: Traditional Methods vs. Rapid Microbial Methods
The following tables summarize the key characteristics and experimental performance data of traditional and rapid methods, highlighting differences in speed, sensitivity, and operational parameters.
Table 1: Overall Method Comparison between Traditional and Rapid Microbial Methods
| Feature | Traditional Methods | Rapid Microbial Methods (RMMs) |
|---|---|---|
| Principle | Culture-based growth on agar plates or in liquid media [1] | Viability-based detection (e.g., ATP bioluminescence, CO2 production, flow cytometry) [51] [52] |
| Time to Result (Sterility Test) | 14 days [50] [7] | 5-8 days, with some positive results detectable in as little as 24-48 hours [52] |
| Time to Result (Bioburden) | 3-5 days for colony formation [51] | Under 45 minutes to a few hours [14] [51] |
| Sensitivity | Detects a wide range of cultivable microbes [1] | High sensitivity; capable of detecting low levels of contamination (e.g., LOQ of 0.1 CFU/mL) and stressed/VBNC organisms [14] [7] |
| Automation & Throughput | Manual, labor-intensive processes [1] | Often automated, reducing human error and increasing throughput [1] [7] |
| Regulatory Status | Well-established and universally accepted gold standard [1] [7] | Gaining regulatory acceptance; requires validation to prove equivalence to traditional methods [7] [52] |
| Key Limitation | Inability to detect Viable But Non-Culturable (VBNC) organisms [7] | High initial investment cost; may not distinguish between viable and non-viable cells (e.g., PCR) [1] [7] |
Table 2: Experimental Performance Data for Select Rapid Sterility Testing Systems
| Performance Parameter | BacT/Alert 3D System (Colorimetric CO2 Detection) | ATP-Bioluminescence System (e.g., Celsis) | Respiration-Based System |
|---|---|---|---|
| Detection Principle | Colorimetric detection of CO₂ produced by microbial growth [50] | Detection of light generated by an enzyme reaction with microbial ATP [52] | Detection of pH change in media caused by microbial respiration [52] |
| Validated Time to Negative Result | Shorter than 14-day compendial method; specific duration not stated [50] | 7 days [52] | 8 days [52] |
| Sample Preparation | Compatible with membrane filtration, allowing for testing of larger sample volumes [50] [52] | Compatible with membrane filtration [52] | Limited to direct inoculation [52] |
| Specificity for Mould | Effectively detects fungi like C. albicans and A. brasiliensis [50] | Validated for broad organism detection, including mould [52] | Reported to have issues detecting mould [52] |
| Automation | Automated, continuous incubation, shaking, and monitoring [50] | Media incubation is performed off-board the instrument [52] | Media incubation occurs on-board the instrument for automated reads [52] |
Experimental Protocols and Methodologies
A valid comparison of methods requires a structured experimental approach based on regulatory guidelines.
Protocol for Validating a Rapid Sterility Test Method
The following workflow outlines the key stages in validating a rapid sterility testing method to demonstrate equivalence to the pharmacopoeial method.
Detailed Validation Steps and Parameters:
Building on the workflow above, the validation process is critical for regulatory acceptance. Key steps and parameters are derived from pharmacopoeial guidelines such as USP <1223> and Ph. Eur. 5.1.6 [14] [7].
- Step 1: Specificity (Growth Promotion Test): This evaluates the ability of the RMM to detect a wide range of relevant microorganisms. The protocol involves inoculating the rapid system with a low number of colony-forming units (CFUs), typically <100 CFU, of challenge organisms. These organisms should include compendial strains like Staphylococcus aureus, Pseudomonas aeruginosa, Bacillus subtilis, Clostridium sporogenes, Candida albicans, and Aspergillus brasiliensis [50]. The RMM must detect these organisms with a performance equivalent or superior to the traditional method.
- Step 2: Limit of Detection (LOD): The LOD is the smallest number of microorganisms that can be reliably detected. Experiments involve testing serial dilutions of microbial suspensions to determine the minimum inoculum, such as 0.25 CFU/5 mL, that the method can consistently detect [50].
- Step 3: Accuracy and Precision: Accuracy measures the closeness of agreement between the RMM and the traditional method. Precision evaluates the agreement among a series of measurements. A study design may include testing ten replicates at low microbial concentrations (e.g., 0.05, 0.1, and 1 CFU/mL) and six replicates at higher concentrations (e.g., 10 and 100 CFU/mL) to calculate metrics like percent recovery and linearity [14].
- Step 4: Robustness and Ruggedness: These tests determine the reliability of the method under small, deliberate variations in operational parameters (e.g., temperature, incubation time) and when performed by different analysts or using different instrument lots [7].
Protocol for a Comparative Bioburden Study
A 2024 study exemplifies a robust protocol for comparing a rapid bioburden analyzer against the traditional plate count method [14].
- Microorganism Preparation: A diverse panel of microorganisms is selected based on pharmacopoeial recommendations and environmental isolates, including bacteria (e.g., B. subtilis, P. aeruginosa, S. aureus) and fungi (e.g., C. albicans, A. brasiliensis). To simulate real-world conditions, microorganisms are starved for three days to create stressed cells [14].
- Sample Preparation and Testing: Serial dilutions of each microorganism are prepared to create a range of concentrations from 0.05 CFU/mL to 100 CFU/mL. Multiple replicates at each concentration are tested in parallel using the rapid method (e.g., Sievers Soleil Rapid Bioburden Analyzer) and the reference traditional method (membrane filtration) [14].
- Data Analysis and Acceptance Criteria: Results from both methods are compared. Acceptance criteria for equivalency often include:
- Linearity: A correlation coefficient (R²) of >0.95 between the two methods.
- Accuracy/Recovery: The recovery of the RMM compared to plate counts should be >50% with a goal of <200% [14].
The Scientist's Toolkit: Key Research Reagent Solutions
Successful implementation and execution of microbiological testing, whether traditional or rapid, depend on specific reagents and materials.
Table 3: Essential Materials for Microbiological Testing
| Item | Function | Example Use-Cases |
|---|---|---|
| Fluid Thioglycollate Medium (FTM) | A general-purpose liquid medium for cultivating aerobic and anaerobic bacteria [50]. | Used in compendial sterility testing as one of the two primary media [50]. |
| Soybean-Casein Digest Medium (SCDM) | A liquid medium for cultivating aerobic bacteria and fungi [50]. | Used in compendial sterility testing as one of the two primary media [50]. |
| BacT/Alert Culture Bottles (e.g., SA, SN) | Culture media bottles with a liquid emulsion sensor for CO₂ detection; used in automated rapid sterility systems [50]. | Used for sterility testing in the BacT/Alert 3D system for aerobic (SA) and anaerobic (SN) microorganisms [50]. |
| Tryptic Soy Agar (TSA) | A general-purpose solid growth medium for the enumeration and cultivation of a wide variety of microorganisms [50] [53]. | Used for bioburden testing via pour plate or membrane filtration methods and for purity confirmation [50] [53]. |
| BioBall SingleShot | A standardized, ready-to-use sample containing a known, low number of specific microorganisms (approx. 30 CFU) [50]. | Used for method validation and growth promotion testing to ensure accurate inoculation levels [50]. |
| VITEK 2 Compact System | An automated system for microbial identification using biochemical test panels [53]. | Used to identify microorganisms isolated from bioburden or environmental monitoring samples [53]. |
The data clearly demonstrate that Rapid Microbiological Methods offer transformative advantages over traditional techniques, primarily through dramatically reduced time-to-results, which enables faster decision-making and product release [14] [51]. While traditional methods remain the regulatory gold standard and are suitable for a broad spectrum of microbes, they are slow and labor-intensive, with a limited ability to detect stressed or VBNC organisms [1] [7].
The choice between methods is not a simple binary. Traditional methods remain vital in many contexts, but the implementation of RMMs is accelerating, driven by regulatory recognition and the compelling need for efficiency and enhanced product safety [7] [52]. For researchers and drug development professionals, the path forward involves a careful evaluation of their specific products, processes, and regulatory landscape. By conducting thorough, structured validations as outlined in this guide, the industry can confidently adopt these advanced technologies, strengthening the overall framework of microbiological quality control.
Overcoming Detection Challenges: Sampling Errors, VBNC States, and Data Interpretation
The accurate detection of microbial contaminants is a cornerstone of pharmaceutical drug development and safety. However, the inherent tendency of bacteria to form aggregates and biofilms presents a formidable challenge to reliable sampling and diagnosis. These complex structures lead to a heterogeneous distribution of pathogens at infection sites or within production environments, making their detection highly dependent on sampling strategy and methodology [54]. This guide objectively compares the performance of traditional culture-based methods with emerging rapid technologies in this critical context. While rapid methods offer significant speed advantages, their success is fundamentally reliant on the initial cultivation step provided by traditional media, a dependency that is acutely accentuated when dealing with biofilms and aggregates [55] [54]. We synthesize current research and experimental data to provide a clear framework for selecting and validating detection strategies that effectively address these sampling complexities.
The Scientific Challenge: Heterogeneity and Detection Limits
Bacterial aggregates and biofilms are structured communities of microorganisms encased in a self-produced extracellular polymeric substance (EPS) matrix. This matrix provides structural integrity and environmental protection, facilitating microbial persistence in adverse conditions [56]. The biofilm life cycle begins with the attachment of planktonic cells to a surface, followed by irreversible adhesion, microcolony formation, and eventual dispersion of cells to new sites [57] [58].
The Critical Impact of Aggregation on Detection Probability
The fundamental challenge in detecting these communities lies in their heterogeneous physical distribution. A 2025 study provides a quantitative model for the probability of detecting bacteria in tissue specimens, which is directly applicable to sampling in bioprocessing environments [54].
The research introduces an aggregation parameter, c, which describes the average size of bacterial aggregates in colony-forming units (CFUs). The probability of obtaining a positive biopsy (or sample) is then modeled by the formula: [ P(at\ least\ one\ positive\ biopsy) \approx 1 - Q\left(1 + \frac{\etal \cdot mB}{c}, \frac{\eta \cdot m_B}{c}\right)^M ] where:
- η is the bacterial load (CFU/g)
- ηℓ is the detection limit (CFU/g)
- m_B is the biopsy/sample size (g)
- M is the number of biopsies/samples [54]
The model's key finding is that cell aggregation has virtually no influence on detection probability when the aggregation parameter is lower than the positive sampling limit (c < ηℓ · m_B). However, once the aggregation parameter exceeds this limit, the probability of detection decreases dramatically [54]. This quantifies the intuitive understanding that larger, more dense aggregates are less likely to be captured in a small sample, leading to false-negative results.
Table 1: Impact of Aggregate Size and Sampling Strategy on Detection Probability
| Aggregate Size (CFU/Aggregate) | Number of Specimens | Probability of Detection | Clinical/Bioprocessing Implication |
|---|---|---|---|
| Small Aggregates (< 1,000 CFU) | 5 | High | Below critical aggregation parameter; multi-specimen strategy effective [54] |
| Large Aggregates (> 1,000 CFU) | 5 | Low | Above critical aggregation parameter; culture-negative diagnoses likely [54] |
| Large Aggregates (> 1,000 CFU) | >5 | Limited Improvement | Increasing specimen count provides diminishing returns [54] |
This model helps explain the high culture-negative rates (up to 20%) reported for conditions like periprosthetic joint infections, where biofilm aggregates are common [54]. The European Bone and Joint Infection Society account for this by defining 2/5 positive tissue samples as a confirmatory criterion for infection, acknowledging the role of heterogeneity [54].
Biofilm Physical Characteristics and Resistance
The physical properties of biofilms further complicate their eradication and detection. Biofilms grown under different conditions exhibit distinct characteristics:
- Low-Shear Biofilms: Are thicker (52 ± 20 µm), rougher, more porous, and less dense, with a higher viscous component in their mechanical properties [59].
- High-Shear Biofilms: Are thinner (29 ± 8 µm), more compact and uniform, and significantly stiffer (creep compliance of 31 ± 1 Pa⁻¹ vs. 5570 ± 101 Pa⁻¹ for low-shear biofilms) [59].
The EPS composition also differs; high-shear biofilms have a three times higher protein-to-polysaccharide ratio in their matrix, suggesting greater cohesion and stability [59]. These physical characteristics directly impact antimicrobial efficacy. For instance, low-shear biofilms showed 80% inactivation when treated with tobramycin combined with low-frequency ultrasound (LFU), whereas high-shear biofilms required higher LFU intensities to achieve similar results, linking mechanical stiffness to treatment resistance [59].
Comparative Analysis of Detection Methods
The Enduring Role of Traditional Culture and Staining
Despite the rise of rapid methods, traditional culture remains the gold standard for detecting viable bacteria in suspected infections due to its cost-effectiveness and ability to provide antimicrobial resistance profiles [54]. Its reliability, however, is contingent on using high-quality culture media that ensures consistent growth, pH, and selectivity batch after batch [55].
Traditional staining methods provide a cost-effective means of visualizing biofilms. The crystal violet assay is a widely used colorimetric method for quantifying total biofilm biomass, though it cannot distinguish between live and dead cells [56]. Similarly, the Congo red agar assay is a qualitative test for extracellular polysaccharide production but offers no information on viability or structure [56]. A novel dual-staining method using Maneval's stain has recently been developed as a simple, cost-effective alternative for light microscopy that differentiates bacterial cells (appearing magenta-red) from the surrounding biofilm matrix (appearing blue) [58].
Table 2: Comparison of Biofilm Detection and Visualization Methods
| Method | Principle | Key Advantages | Key Limitations | Distinguishes Cells from Matrix? |
|---|---|---|---|---|
| Traditional Culture | Growth on solid or liquid media | Gold standard for viability; allows AMR profiling [54] | Time-consuming (up to 14 days); cannot detect VBNC state [54] | No |
| Crystal Violet Staining | Dye binding to cells and matrix [56] | Simple, cost-effective, high-throughput [56] | Does not distinguish viable/dead cells; no structural data [56] | No [58] |
| Dual-Staining (Maneval's) | Sequential Congo red and Maneval's staining [58] | Cost-effective; works with basic light microscope; differentiates cells from matrix [58] | Less quantitative than advanced methods | Yes [58] |
| Label-Free Analysis (LFAB) | Brightfield microscopy with OD measurement [60] | Non-perturbative, real-time monitoring, high-throughput [60] | Requires specialized imaging setup and analysis software | Yes, via optical contrast |
| Confocal Laser Scanning Microscopy (CLSM) | Fluorescence optical sectioning | High-resolution 3D architecture; cell viability staining [57] | Requires fluorescent probes; expensive; complex [60] [58] | Yes, with specific stains |
Emerging and Rapid Methodologies
Emerging technologies are enhancing our ability to study and detect biofilms with greater speed and resolution.
- Label-Free Analysis of Biofilms (LFAB): This imaging approach combines time-lapse, low-magnification brightfield microscopy with regional optical density measurements to quantify biofilm biomass in real-time without dyes or labels [60]. It is non-perturbative and allows for high-temporal-resolution monitoring of biofilm growth dynamics, correlating strongly with traditional assays like crystal violet and confocal microscopy [60].
- Advanced Imaging and Sensing: Techniques like atomic force microscopy (AFM) provide nanomechanical data on biofilm adhesion and elasticity [57]. Microfluidic platforms simulate relevant environments to study biofilm heterogeneity and antimicrobial responses under controlled conditions [57]. Biosensors are also being developed for real-time, on-site pathogen detection with high sensitivity [61].
- Molecular Methods: Quantitative PCR (qPCR) and Next-Generation Sequencing (NGS) rapidly detect and quantify biofilm-related genes and microbial populations [57]. CRISPR-based technologies are being explored for both targeted gene editing and the development of highly specific biosensing systems for detecting biofilm-associated genes [57].
Experimental Protocols for Key Studies
Protocol: Dual-Staining Method for Biofilm Visualization
This protocol, adapted from Singh et al. (2024), details a cost-effective method for visualizing and differentiating biofilm components [58].
- Biofilm Growth: Grow biofilm on a sterile glass slide submerged in nutrient broth inoculated with a 1:100 dilution of a 0.5 McFarland standard bacterial suspension. Incubate undisturbed at 37°C for 3 days [58].
- Rinsing and Fixation: Gently rinse the slide by dipping it in distilled water for 5 seconds to remove non-adhered cells. Fix the biofilm by immersing the slide in 4% formaldehyde (in distilled water) for 15-30 minutes at room temperature [58].
- Staining: a. Treat the fixed biofilm with 1% Congo red stain and allow it to air-dry completely [58]. b. Subsequently, cover the biofilm with Maneval's stain for 10 minutes [58]. c. Remove excess stain and air-dry the slide [58].
- Visualization: Observe the slide under a light microscope using a 100x oil immersion objective. Bacterial cells appear magenta-red, surrounded by a blue-stained polysaccharide biofilm matrix [58].
Protocol: Label-Free Analysis of Biofilms (LFAB)
This protocol, based on Blanken et al. (2025 preprint), describes a non-invasive method for quantifying biofilm microcolonies [60].
- Inoculation and Imaging: Inoculate dilute bacterial cultures (e.g., 10⁴ cells/mL) into glass-bottom microtiter dishes. Perform time-lapse imaging of static culture growth using a 4x or 10x magnification objective on a brightfield microscope [60].
- Image Analysis Pipeline: a. Background Subtraction: Apply a local background subtraction to raw images to correct for illumination unevenness [60]. b. Thresholding and Mask Creation: Blur the image to reduce internal intensity variation and apply a constant intensity threshold to create a binary mask that identifies biofilm regions [60]. c. Optical Density Conversion: Convert pixel-wise intensity values in the raw images to optical density (OD) values using a log10-based transformation that accounts for maximum transmittance (a blank control) and camera noise [60]. d. Biomass Calculation: Apply the binary biofilm mask to the OD image. The average optical density of the segmented biofilm image is reported as the "BF-biofilm biomass," a metric that accounts for both biofilm coverage and density [60].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Reagents and Materials for Biofilm Detection Studies
| Item | Function/Application | Example Use Case |
|---|---|---|
| Redipor Prepared Media (e.g., TSA, TSB) [55] | Provides consistent, high-quality microbial growth foundation for both traditional and rapid methods. | Culturing colonies from homogenized tissue samples prior to MALDI-TOF identification [55]. |
| Maneval's Stain [58] | A key component in the dual-staining method for differential visualization of cells (magenta-red) and matrix (blue). | Cost-effective, routine visualization and confirmation of biofilm formation in clinical isolates using basic light microscopy [58]. |
| Congo Red Stain [56] [58] | Dye used for qualitative assessment of exopolysaccharide production on agar or as part of staining protocols. | Preliminary screening of bacterial strains for slime production; first step in the dual-staining method [56] [58]. |
| Polystyrene or Glass-Bottom Microtiter Dishes [60] | Standard substrate for growing biofilms in vitro under static conditions for various assays. | Performing crystal violet assays, LFAB imaging, or screening antimicrobial agents against biofilms [60] [56]. |
| Low-Frequency Ultrasound (LFU) Device [59] | Used as an adjuvant to enhance antibiotic penetration and efficacy against biofilms in research settings. | Studying mechanisms of biofilm disruption and modeling combination therapies for resistant infections [59]. |
| Reference Microbial Strains (e.g., ATCC standards) [62] | Essential controls for validating detection methods, ensuring accuracy, and meeting regulatory guidelines. | Method validation and quality control during routine testing of biopharmaceutical products and raw materials [62]. |
Visualizing Workflows and Relationships
Biofilm Detection Decision Workflow
The following diagram outlines a logical pathway for choosing an appropriate biofilm detection method based on research objectives and resource constraints.
Sampling and Detection Challenge
This diagram illustrates the core problem of sampling heterogeneous bacterial aggregates, as modeled in recent research [54].
The effective detection of bacterial aggregates and biofilms requires a nuanced strategy that acknowledges the critical interplay between sampling, traditional methods, and rapid technologies. The quantitative model demonstrating the severe impact of aggregation size on detection probability provides a scientific basis for optimizing sampling plans [54]. While rapid identification methods like MALDI-TOF MS and molecular assays have transformed the pace of diagnostics, they remain fundamentally dependent on the initial, reliable growth provided by traditional culture media [55]. Therefore, the future of microbial detection in complex matrices lies not in a binary choice between traditional and rapid methods, but in their integrated application. This includes using traditional media for robust cultivation, leveraging new, cost-effective staining methods for visualization, and adopting high-throughput, label-free imaging for dynamic studies. A comprehensive, proactive contamination control strategy that understands and addresses these sampling complexities is essential for ensuring product safety in drug development and accurate diagnosis in clinical settings.
The Viable But Non-Culturable (VBNC) state represents a dormant survival mechanism employed by many bacteria when facing environmental stress. In this state, microorganisms fail to grow on conventional culture media—the gold standard of traditional microbiology—while maintaining metabolic activity and the potential to resuscitate under favorable conditions [63] [64]. For pharmaceutical researchers and drug development professionals, this phenomenon presents a significant challenge: traditional growth-based methods may underestimate microbial contamination, potentially compromising product safety and process control.
The transition to the VBNC state can be induced by various stressors common in pharmaceutical manufacturing environments, including nutrient starvation, extreme temperatures, osmotic pressure, and exposure to disinfectants or antibiotics [63] [65] [66]. Over 100 bacterial species, including pathogens highly relevant to pharmaceutical quality control such as Escherichia coli, Salmonella enterica, Staphylococcus aureus, and Listeria monocytogenes, have been documented to enter this resilient state [63] [65]. This survival strategy allows bacteria to persist through manufacturing processes and evade detection by conventional microbiological monitoring, creating a hidden risk that Rapid Microbiological Methods (RMM) are uniquely positioned to address.
Understanding the VBNC State: Characteristics and Formation Mechanisms
Defining Characteristics of VBNC Organisms
VBNC cells exhibit three fundamental characteristics that distinguish them from both culturable and dead cells. First, they cannot form colonies on standard growth media typically used in quality control laboratories, leading to false-negative results in traditional tests [63] [64]. Second, they maintain viability and metabolic activity, as demonstrated through various activity stains and molecular methods [63] [64]. Third, they retain the capacity to resuscitate when favorable conditions return, potentially regaining full culturalility and pathogenicity [63] [67] [64].
Morphologically, VBNC cells typically undergo reduction in size—often described as "rounding up" in Vibrio species—and may become "ultramicrobacteria" capable of passing through 0.2μm filters nominally rated as sterilizing grade [66]. Physiologically, they demonstrate markedly reduced metabolic rates, lower respiration, reduced nutrient transport, and minimal synthesis of macromolecules, enabling survival for extended periods exceeding one year in some cases [63].
Environmental Triggers and Molecular Mechanisms
The transition to the VBNC state occurs in response to adverse conditions regularly encountered in pharmaceutical manufacturing and control environments. Table 1 summarizes the primary environmental triggers and their relevance to pharmaceutical contexts.
Table 1: Environmental Triggers for VBNC State Formation in Pharmaceutical Contexts
| Inducing Stress | Examples in Pharmaceutical Context | Representative Affected Organisms |
|---|---|---|
| Nutrient starvation | Low-nutrient water systems (PW, WFI), ultra-pure environments | Staphylococcus aureus, Escherichia coli [65] |
| Extreme temperatures | Cold storage, freeze-thaw cycles, thermal processing | Vibrio parahaemolyticus, Campylobacter jejuni [65] |
| Disinfectant exposure | Hypochlorite, hydrogen peroxide, quaternary ammonium compounds | Legionella pneumophila, Listeria monocytogenes [67] [66] |
| Osmotic pressure | High-salt buffers, syrup formulations | Proteus mirabilis, Vibrio vulnificus [65] |
| pH extremes | Acidic/basic cleaning agents, product formulations | Escherichia coli O157:H7 [65] |
| Oxidative stress | Peracetic acid, hydrogen peroxide sterilization | Salmonella enterica [65] |
At the molecular level, the transition involves sophisticated genetic regulation. Two key mechanisms have been identified:
Stringent Response: This survival mechanism is triggered through synthesis of the alarmone (p)ppGpp, which reconfigures cellular processes by binding to targets like RNA polymerase, affecting replication, transcription, and translation. Elevated activity of the relA and spoT genes leads to (p)ppGpp accumulation, enhancing stress resistance and facilitating entry into dormancy [65].
Toxin-Antitoxin (TA) System: This system involves stable toxin proteins that slow bacterial growth and unstable antitoxin proteins that inhibit toxin expression. Under stress conditions, activated toxin proteins mediate physiological changes that promote dormancy, including reduced metabolism and cell division [65].
Figure 1: VBNC State Induction Pathway and Characteristics
Methodological Comparison: Traditional Culture vs. Rapid Microbiological Methods
Limitations of Traditional Culture-Based Methods
Traditional microbiological methods, which rely on the ability of microorganisms to proliferate in or on culture media, form the basis of current compendial tests for pharmaceutical quality control. These include agar plating, membrane filtration, and broth enrichment techniques prescribed for raw material testing, bioburden assessment, environmental monitoring, and sterility testing [66]. While these methods have served as the industry standard for decades, they possess fundamental limitations in detecting VBNC organisms.
The primary drawback is their dependence on cellular division and colony formation as indicators of viability. Since VBNC cells are metabolically active but non-dividing, they cannot be detected by these growth-based methods, creating a significant detection gap [67] [66]. This limitation is particularly concerning for pharmaceutical water systems, where VBNC organisms are prevalent and may include pathogenic species that retain virulence despite their non-culturable status [66]. Additionally, traditional methods are time-intensive, requiring several days to obtain results (e.g., 5-6 days for enterococci detection), which delays decision-making and product release [68].
Advantages of Rapid Microbiological Methods for VBNC Detection
Rapid Microbiological Methods (RMM) encompass technologies that detect microorganisms through mechanisms other than growth-based culture. These methods target various cellular components, activities, or byproducts as indicators of viability, making them uniquely suited for detecting VBNC organisms [8] [69]. The fundamental advantage of RMM lies in their ability to detect metabolic activity or cellular components independent of replication, allowing for identification of VBNC cells that remain undetectable by traditional means [8].
Different RMM technologies offer distinct approaches to overcoming the VBNC detection challenge. Table 2 compares the operational characteristics and capabilities of major RMM categories against traditional methods.
Table 2: Comparison of Microbial Detection Methods for VBNC Organisms
| Method Category | Specific Technologies | Detection Principle | Time to Result | VBNC Detection Capability | Key Limitations |
|---|---|---|---|---|---|
| Traditional Culture | Agar plating, broth enrichment, membrane filtration | Cellular growth and division | Days to weeks | No | Labor-intensive, subjective, misses VBNC cells [68] [8] [66] |
| Nucleic Acid-Based | qPCR, ddPCR with viability markers (PMA) | Detection of DNA/RNA from membrane-intact cells | Hours | Yes | Requires sample preprocessing, may not distinguish viability at low metabolic states [70] |
| Viability Staining & Flow Cytometry | Fluorescent viability stains, solid phase cytometry | Membrane integrity, enzymatic activity | Minutes to hours | Yes | May require specialized expertise, equipment cost [64] [69] |
| Bioluminescence | ATP bioluminescence | Detection of cellular ATP | Minutes | Yes (with limitations) | Variable ATP content between species, interference from cleaning agents [8] [69] |
| Impedance & Conductance | Impedance microbiology | Metabolic conversion changing electrical properties | Hours to days | Partial | Limited sensitivity, medium-dependent [68] [8] |
| Automated Colony Detection | Autofluorescence detection with optics/camera | Colony formation without visual inspection | Days (reduced labor) | No | Still growth-dependent, similar timeframes to traditional methods [69] |
As noted in recent industry guidance, "MMM includes technologies based on the use of intrinsic fluorescence, extrinsic fluorescence (e.g., viability staining), bioluminescence, enzyme indicators, Raman spectroscopy, flow cytometry, solid phase cytometry, polymerase chain reaction (PCR) and automated colony detection and counting" [69]. These methods can offer "a shorter time to detection, real-time reporting of results, continuous monitoring, higher sensitivity and a lower false negative rate (e.g., due to detection of viable but not culturable)" compared to traditional approaches [69].
Experimental Evidence: Comparative Studies and Validation Data
Direct Method Comparison Studies
Industry trials and comparative studies provide compelling experimental evidence supporting the advantage of RMM for detecting VBNC organisms. In one comprehensive study comparing traditional and rapid methods for monitoring surface hygiene in food processing environments (with direct relevance to pharmaceutical manufacturing), researchers found significant differences in detection capabilities between methods [71]. The study demonstrated that each testing method passed or failed different surfaces, suggesting complementary detection profiles and highlighting the limitations of relying exclusively on traditional culture-based approaches.
A specific comparison between standard and impedance methods for detecting enterococci contamination in medicinal herbs revealed important practical insights. While both methods showed general agreement in overall contamination rates (33.7% for standard vs. 25% for impedance method), the impedance method provided results in 0.66-21.11 hours compared to 5-6 days required for the standard method [68]. This dramatic reduction in time-to-result represents a significant operational advantage while maintaining detection reliability.
Advanced Molecular Detection Protocols
Recent advances in molecular biology have yielded increasingly sophisticated methods for VBNC detection and quantification. A 2025 study developed an optimized protocol for absolute quantification of VBNC state formation and resuscitation in Klebsiella pneumoniae using propidium monoazide droplet digital PCR (PMA-ddPCR) [70]. This method enables direct quantification of viable cells without requiring external standard curves, providing unprecedented accuracy in VBNC cell enumeration.
The experimental protocol involves several critical steps:
- PMA treatment optimization: Testing concentrations between 5μM and 200μM with incubation times from 5-30 minutes to selectively penetrate membrane-compromised cells
- DNA extraction: Standardized isolation of genetic material from both viable and non-viable cells
- Droplet digital PCR: Partitioning of samples into thousands of nanoliter-sized droplets for absolute quantification of target genes without standard curves
- Viability assessment: Comparison of signal reduction between treated and untreated samples to determine viable cell counts [70]
This approach demonstrated detection sensitivity capable of measuring activity reductions ranging from 1.13 to 0.64 log10 DNA copies/mL in complex matrices like mouse fecal samples, highlighting its potential for pharmaceutical water system monitoring and contamination investigation [70].
Figure 2: PMA-ddPCR Workflow for VBNC Cell Quantification
The Scientist's Toolkit: Essential Research Reagents and Solutions
Effective detection and study of VBNC organisms requires specialized reagents and materials designed to differentiate viable cells based on various physiological criteria rather than growth capacity. Table 3 catalogues key research solutions essential for investigating the VBNC state.
Table 3: Essential Research Reagents for VBNC Detection and Analysis
| Reagent Category | Specific Examples | Function/Application | Detection Target |
|---|---|---|---|
| Viability Stains | SYTO-9, propidium iodide (BacLight Kit), CTC, fluorescein diacetate | Membrane integrity assessment, metabolic activity detection | Membrane potential, enzyme activity, respiration [64] [66] |
| Nucleic Acid Viability Markers | Propidium monoazide (PMA), PMAxx, ethidium monoazide | Selective DNA amplification from viable cells with intact membranes | Membrane-intact cells (excludes free DNA and compromised cells) [70] |
| Metabolic Indicators | Tetrazolium salts (XTT, MTT), resazurin | Detection of cellular reductase activity | Electron transport chain activity, cellular reduction capacity [64] |
| Molecular Detection Reagents | PCR master mixes, specific primers/probes, DNA extraction kits | Genetic detection and quantification of viable pathogens | Species-specific gene targets (e.g., rpoB, adhE) [70] |
| Enrichment Media Components | Bromocresol Purple Azide broth, Kanamycin Esculin Azide broth | Selective enrichment of target organisms before detection | Growth recovery of injured but culturable cells [68] |
| Impedance Media | BiMedia 330A, other specialized conductivity media | Detection of metabolic conversion through electrical changes | Metabolic byproducts that alter media conductivity [68] |
These reagents enable researchers to implement the methodologies discussed throughout this article, from simple viability staining to sophisticated molecular detection protocols. When establishing a new detection method, it is essential to systematically optimize critical parameters such as dye concentrations, incubation conditions, and inhibitor concentrations to ensure accurate differentiation between viable, VBNC, and non-viable cell populations.
Implications for Pharmaceutical Quality Control and Contamination Control Strategy
The detection of VBNC organisms has profound implications for pharmaceutical quality systems and contamination control strategies. Regulatory guidance, including the revised EU GMP Annex 1, now explicitly emphasizes the importance of modern microbial methods as part of a comprehensive Contamination Control Strategy (CCS) [69]. This regulatory evolution reflects growing recognition that traditional methods alone may not provide sufficient protection against microbial risks, particularly those posed by VBNC organisms.
Implementation of RMM for VBNC detection offers tangible benefits across multiple pharmaceutical quality systems:
Water System Control: Detection of VBNC organisms in purified water and WFI systems provides a more accurate assessment of microbial contamination risks, enabling more effective sanitization regimen design and system control [66] [69].
Environmental Monitoring: Advanced methods like flow cytometry and solid phase cytometry can detect VBNC populations in cleanrooms and manufacturing environments, offering enhanced insight into microbial recovery after sanitization [69].
Raw Material Testing: Implementation of rapid methods sensitive to VBNC states allows for more comprehensive risk assessment of incoming materials, particularly those of natural origin that may harbor stressed microorganisms [66] [69].
Process Validation: Understanding the true bioburden, including VBNC organisms, enables more accurate challenge studies and process design, particularly for non-sterile products where microbial control is critical to quality [66].
While some have questioned the direct patient risk from VBNC organisms due to their limited replication capacity, the potential for resuscitation and regained pathogenicity in pharmaceutical products represents a risk that cannot be ignored [67] [66]. As the industry moves toward more robust contamination control strategies, the ability to detect VBNC organisms through rapid microbiological methods will become increasingly integral to comprehensive quality systems.
The limitation of traditional, growth-based microbiological methods in detecting VBNC organisms represents a significant gap in pharmaceutical quality control. Rapid Microbiological Methods overcome this limitation through diverse technological approaches that detect viability independent of culturability, providing a more comprehensive assessment of microbial contamination risks. As the pharmaceutical industry advances toward real-time release testing and more sophisticated contamination control strategies, the implementation of RMM capable of VBNC detection will transition from a competitive advantage to an industry essential. The experimental evidence and methodological comparisons presented in this article demonstrate that RMM technologies offer not just rapid results, but fundamentally more accurate detection of viable microorganisms—including those in the problematic VBNC state—thereby strengthening the foundation of pharmaceutical product quality and patient safety.
Microbiological testing stands as a critical gatekeeper for ensuring product safety and quality across pharmaceutical, food, and beverage industries. For decades, traditional culture-based methods have served as the gold standard, relying on the growth of microorganisms on artificial media to detect contamination. While these methods are well-established and reliable, their significant limitation lies in the extended incubation period of 24 hours to 14 days required to obtain results, creating operational bottlenecks [1] [22]. In response to the pressing need for faster feedback, rapid microbiological methods (RMMs) have emerged as powerful alternatives. These innovative techniques leverage advanced technologies to dramatically reduce the time-to-result (TTR), in many cases providing data within hours or even minutes [1] [72]. However, the selection between traditional and rapid methods is not straightforward, often involving a strategic trade-off between speed and analytical sensitivity. This guide provides a comparative evaluation of these methodologies, offering researchers and drug development professionals a framework for strategic method selection based on empirical data and application-specific requirements.
Comparative Analysis of Method Categories
The evolution of microbiological testing has produced a diverse toolkit of methods, each with distinct operational profiles. The following table provides a high-level comparison of the main methodological categories.
Table 1: Overview of Major Microbiological Testing Methodologies
| Method Category | Key Examples | Typical Time-to-Result | Key Advantages | Primary Limitations |
|---|---|---|---|---|
| Traditional Methods | Plate Count, Most Probable Number (MPN) | 48 - 72 hours (up to 14 days for sterility) [1] | Well-established, regulatory acceptance, detects viable cells, low equipment cost [1] | Time-consuming, labor-intensive, cannot detect VBNC state [1] [73] |
| Growth-based RMM | ATP Bioluminescence, Autofluorescence, Colorimetric Detection | 24 - 48 hours (often includes enrichment) [72] | Faster than traditional methods, mirrors cultural method, easier validation [72] | May require enrichment, limited to cultivable organisms |
| Viability-based RMM | Flow Cytometry, Solid-Phase Cytometry | Minutes to hours after sample processing [73] | Very rapid, detects viable cells (including VBNC), no enrichment needed [73] | High equipment cost, may require specialized training |
| Molecular Methods | PCR, qPCR, ELISA, Biosensors | Several hours [1] [11] | High sensitivity & specificity, can target specific pathogens, amenable to automation [1] | May not distinguish live/dead cells, high initial investment, regulatory hurdles [1] |
To aid in the strategic selection process, the following diagram visualizes the relationship between key decision factors.
Figure 1: A decision pathway for selecting microbiological testing methods based on key project requirements such as speed, regulatory needs, target specificity, and the need for viability assessment.
Experimental Data: Quantitative Performance Comparison
Empirical data is crucial for understanding the practical performance of different methods. The following tables summarize key quantitative findings from validation studies and recent research.
Table 2: Performance Comparison of a Rapid Bioburden Analyzer vs. Traditional Plate Count
| Performance Parameter | Sievers Soleil Rapid Bioburden Analyzer | Traditional Membrane Filtration |
|---|---|---|
| Time-to-Result | < 45 minutes [14] | 48 - 72 hours [14] |
| Limit of Detection (LOD) | 0.05 CFU/mL [14] | Dependent on volume filtered |
| Limit of Quantification (LOQ) | 0.1 CFU/mL [14] | Typically 1 CFU/filter |
| Average Percent Recovery | 140.9% (Goal: 50-200%) [14] | 100% (Reference method) |
| Linearity (R²) | > 0.95 across 3-4 logs [14] | Not specified |
Table 3: Validation Study Results for a Modified Rapid AST Protocol (6-h vs. 24-h Incubation)
| Organism Group (No. of isolates) | Total Comparisons | Interpretive Agreement | Average Zone Size Difference |
|---|---|---|---|
| Staphylococcus spp. (21) | Not Specified | 99.65% (855/858) [74] | 1.08 mm (SD: 1.33 mm) [74] |
| Enterobacterales (20) | Not Specified | 99.65% (855/858) [74] | 1.08 mm (SD: 1.33 mm) [74] |
| Enterococcus/Streptococcus spp. (21) | Not Specified | 99.65% (855/858) [74] | 1.08 mm (SD: 1.33 mm) [74] |
| Haemophilus influenzae/ Moraxella catarrhalis (20) | Not Specified | 99.65% (855/858) [74] | 1.08 mm (SD: 1.33 mm) [74] |
Detailed Experimental Protocols
To ensure reproducibility and provide insight into experimental rigor, this section outlines key protocols cited in the comparative data.
Protocol 1: Validation of a Rapid Bioburden Analyzer
This protocol, adapted from a study evaluating the Sievers Soleil analyzer, details the process for demonstrating equivalency to the traditional plate count method [14].
- Microorganism Preparation: Select microorganisms based on pharmacopoeial recommendations (e.g., A. brasiliensis, B. subtilis, E. coli, P. aeruginosa, S. aureus, C. albicans, etc.). Prepare stock solutions and subject them to starving conditions for three days to simulate real-world, stressed organisms [14].
- Sample Dilution and Inoculation: Create serial dilutions of each stock solution to achieve concentrations between 0.05 CFU/mL and 100 CFU/mL. Inoculate these into a defined matrix, such as Water For Cell Culture. Aliquot samples for parallel testing on the rapid analyzer and via membrane filtration [14].
- Parallel Testing & Data Analysis:
- Rapid Method: Analyze samples according to the manufacturer's instructions. The system detects and quantifies microbial loads automatically.
- Reference Method: Filter aliquots through a membrane, place on agar, and incubate for the standard duration (e.g., 48-72 hours). Count resulting Colony Forming Units (CFUs).
- Compare results from both methods to calculate percent recovery, linearity, and limits of detection/quantification. The acceptance criteria for recovery is typically >50% with a goal of <200%, and linearity (R²) should be >0.95 [14].
The workflow for this comparative validation is illustrated below.
Figure 2: A workflow for the validation of a rapid bioburden analyzer against the traditional membrane filtration method, highlighting the parallel testing pathway.
Protocol 2: Rapid Antimicrobial Susceptibility Testing (AST) Direct from Blood Culture
This protocol describes a study that modified the standard EUCAST disk diffusion method to reduce turnaround time for positive blood cultures, a critical scenario in clinical diagnostics [74].
- Sample Collection and Inoculation: Collect positive blood culture samples. Instead of the standard subculture incubation of 16-24 hours, directly inoculate Mueller Hinton Agar (MHA) plates from the positive blood culture bottle after initial organism identification (e.g., by MALDI-TOF) [74].
- Short-incubation AST: Incubate the inoculated AST plates for only 6 hours at the standard temperature (e.g., 35°C ± 2°C) [74].
- Reading and Comparison: After the 6-hour incubation, measure the zones of inhibition for the antibiotic disks. Interpret the results as Sensitive (S), Resistant (R), or Intermediate (I) using EUCAST guidelines. In parallel, prepare and incubate standard 24-hour AST cultures from the same sample. Compare the qualitative interpretive categories and the quantitative zone diameters from the 6-hour and 24-hour plates to determine the agreement rate and any significant differences [74].
Application-Specific Considerations for the Pharmaceutical Industry
The choice between traditional and rapid methods is highly contextual. Within the pharmaceutical industry, specific applications present unique requirements that influence the optimal strategy.
- Sterility Testing and Product Release: For final product release, regulatory compliance is paramount. Traditional methods remain the official compendial method. However, RMMs like autofluorescence detection (e.g., Growth Direct System) can cut TTR by approximately half while closely mirroring the traditional culture process, facilitating validation and regulatory submission [72]. Their implementation can significantly reduce inventory holding costs and enable more rapid release of products with short shelf-lives.
- Environmental Monitoring (EM): Rapid air samplers, such as the Coriolisμ, collect microbes in liquid for subsequent rapid analysis (e.g., PCR), providing faster data on cleanroom state [72]. Real-time systems like Instantaneous Microbial Detection (IMD) using Mie scattering and intrinsic fluorescence can provide immediate alerts to contamination events, allowing for proactive intervention, which is a powerful tool for Process Analytical Technology (PAT) [72].
- Water System Bioburden Control: Monitoring pharmaceutical water systems is a frequent activity. Rapid methods like ATP-bioluminescence or the specialized bioburden analyzers can provide results in minutes to hours, enabling near-real-time system control and faster investigation of microbial excursions compared to the 48-72 hour wait associated with traditional plate counts [72] [14].
- Raw Material and In-Process Testing: The high volume and need for speed in these areas make them ideal for RMM implementation. Faster results on raw materials can reduce quarantine times, while in-process testing allows for more responsive process adjustments, aligning with the FDA's PAT initiative and a "Quality by Design" framework [72].
The Scientist's Toolkit: Key Reagent Solutions
Successful implementation of microbiological testing, whether traditional or rapid, relies on a foundation of specific reagents and materials.
Table 4: Essential Research Reagents and Materials for Microbiological Testing
| Reagent/Material | Function/Application | Key Considerations |
|---|---|---|
| Selective & Non-selective Culture Media | Supports growth and differentiation of microorganisms in traditional and growth-based RMMs. | Choice depends on target organisms; must be validated for intended use and meet fertility requirements [22]. |
| Adenosine Triphosphate (ATP) Reagents | Core component of ATP-bioluminescence assays. Luciferin/luciferase enzyme reaction produces light proportional to microbial ATP [72]. | Requires steps to eliminate non-microbial ATP from the sample, which can cause interference [72]. |
| Specific Primers and Probes | Essential for molecular methods (PCR, qPCR) to target and amplify unique nucleic acid sequences of pathogens or indicators [11]. | Design is critical for specificity and sensitivity; must be validated against a panel of relevant organisms. |
| Antibiotic Disks | Used in Antibiotic Susceptibility Testing (AST) by disk diffusion to determine microbial resistance profiles [74]. | Potency and stability are critical; must be stored appropriately and used within expiry dates. |
| Viability Stains (e.g., for Flow Cytometry) | Fluorescent dyes that distinguish live/dead cells based on membrane integrity, used in viability-based RMMs [73]. | Dye selection and staining protocol must be optimized for the sample matrix and target microbes. |
| Validation Strains (e.g., ATCC Strains) | Well-characterized microorganisms used for method qualification, validation, and ongoing quality control. | Should include a panel of representative organisms (Gram-positive, Gram-negative, yeast, mold) relevant to the test and sample type [14]. |
The paradigm of microbiological testing is shifting from a one-size-fits-all approach to a strategic, application-driven selection process. Traditional culture methods remain indispensable for their proven accuracy, regulatory acceptance, and ability to provide a viable isolate for further characterization [1] [75]. Conversely, Rapid Microbiological Methods offer transformative benefits in speed, sensitivity, and potential for automation, which are critical for modern manufacturing and clinical diagnostics [1] [72].
The experimental data presented confirms that well-validated RMMs can provide equivalent or superior performance to traditional methods for specific applications, such as bioburden testing and AST, while drastically reducing turnaround time [74] [14]. The optimal strategy is not a simple replacement but a thoughtful integration. Researchers and quality control professionals must balance the factors of speed, sensitivity, regulatory requirements, and cost to select the most effective method for their specific context. As regulatory frameworks continue to evolve and embrace modernized approaches like PAT, the strategic adoption of RMMs will undoubtedly play a pivotal role in enhancing product safety, optimizing processes, and accelerating development timelines.
Navigating High Initial Investment and Cost-Benefit Analysis for RMMs
The adoption of Rapid Microbiological Methods (RMMs) represents a significant technological advancement over traditional, growth-based techniques for contamination detection in pharmaceutical manufacturing and research. While RMMs offer compelling technical benefits—including dramatically reduced time-to-result, enhanced sensitivity, and greater automation—their implementation is often hampered by a substantial financial barrier. The high initial investment required for RMM instrumentation, validation, and training creates a complex decision-making scenario for researchers, scientists, and drug development professionals. This analysis objectively compares the performance and financial aspects of RMMs against traditional methods, providing a structured framework for navigating the cost-benefit landscape. By synthesizing current experimental data, financial models, and implementation protocols, this guide aims to equip scientific professionals with the evidence needed to justify RMM adoption based on both operational superiority and economic viability.
The pharmaceutical industry's hesitancy to adopt RMMs primarily stems not from technical or regulatory concerns, but from a lack of understanding of how to assess the implementation costs and build a compelling financial case [76]. A comprehensive cost-benefit analysis must extend beyond simple capital expenditure to include operational savings, cost avoidances, and qualitative improvements in quality control and risk management. This guide provides the analytical tools and experimental data necessary to transform RMM evaluation from a financial dilemma into a strategic investment decision.
Understanding the Cost Structure of RMMs
Composition of Initial Investment
The initial investment for RMM implementation is multifaceted, extending beyond the purchase price of instrumentation. A comprehensive financial analysis must account for several interconnected cost components that collectively define the total barrier to entry. These include the capital costs for specialized equipment, which can range from several thousand to hundreds of thousands of pounds depending on system complexity [7]. Additionally, organizations must budget for system qualification and method validation activities, which ensure regulatory compliance but require significant resource allocation. Technology and software training represents another crucial investment, as laboratory personnel must develop new competencies to operate advanced instrumentation effectively. Finally, regulatory filing costs may be necessary if the RMM implementation involves changes to methods specified in existing marketing authorizations [77] [76].
The substantial variance in implementation costs reflects the diversity of RMM technologies available. Techniques such as nucleic acid-based testing (e.g., PCR, qPCR), viability-based assays (e.g., flow cytometry), and cellular component-based techniques (e.g., endotoxin testing) each carry distinct investment profiles [78]. For instance, MALDI-TOF systems offer rapid identification with relatively low per-sample costs but require a significant initial outlay that may be prohibitive for smaller laboratories [7]. This heterogeneity in cost structures necessitates technology-specific financial analysis tailored to the organization's testing volume and application requirements.
Operational Cost Components
Beyond initial investment, a complete financial analysis must consider the ongoing operational costs associated with both traditional methods and RMMs. For conventional growth-based methods, these include recurring expenses for consumables, reagents, and supplies such as culture media and agar plates. The cost of labor represents a particularly significant component, as traditional methods demand extensive hands-on time for sample preparation, incubation monitoring, and manual colony counting [79]. Additional operational expenses include media, reagents, and consumables disposal costs; laboratory equipment depreciation, calibration, and qualification; overhead for laboratory and storage space; data management and record retention; and preventive maintenance and service contracts [77].
RMMs fundamentally reshape this operational cost structure, typically reducing labor requirements and consumables usage while potentially increasing costs for specialized reagents and technical support. This cost structure transformation is a critical consideration in financial modeling, as the balance between capital investment and operational expenditure shifts dramatically with RMM adoption.
Performance Comparison: RMMs vs. Traditional Methods
Experimental Data on Method Performance
Table 1: Comparative Performance of Microbiological Methods
| Parameter | Traditional Cultural Methods | Rapid Microbiological Methods | Experimental Measurement |
|---|---|---|---|
| Time-to-Result | 3-14 days (depending on application) [79] | Hours to 2 days [78] | Incubation period vs. real-time detection |
| Sensitivity | Limited to ~102-103 CFU/mL [80] | Single-cell detection possible [76] | Limit of Detection (LOD) studies |
| Detection of VBNC States | Generally unable to detect [80] | Enhanced detection capabilities [76] | Comparison of viability markers vs. growth |
| Accuracy & Reproducibility | Subject to human error in interpretation [79] | Automated, objective reading [79] | Statistical comparison of replicate samples |
| Sample Throughput | Limited by manual processing | High throughput with automation [78] | Samples processed per unit time |
Independent validation studies demonstrate that RMMs consistently outperform traditional methods across critical performance parameters. A 2020 study investigating 3M Petrifilm for dietary supplement testing found recoveries of 79-123% across multiple product categories, successfully meeting the acceptance criterion of >70% recovery compared to control methods [15]. This demonstrates that properly validated RMMs can deliver equivalent or superior accuracy while providing results in a fraction of the time required by traditional approaches. The technical benefits extend beyond speed to include greater accuracy, precision, sensitivity, and reproducibility, enabling single-cell detection and enhanced identification of stressed and viable but non-culturable (VBNC) organisms that conventional methods often miss [76].
Limitations of Traditional Methods
Traditional growth-based methods suffer from several inherent limitations that impact both operational efficiency and contamination control effectiveness. The most significant constraint is the extensive time required, as microbes need days to weeks to grow sufficiently to be visible to the human eye [79]. This delay creates a critical gap between sampling and result availability, preventing timely intervention during manufacturing processes. Traditional methods also demonstrate limited accuracy because colonies must grow to millions of cells before detection, and even experienced technicians can miscount, potentially leading to unnecessary investigations or undetected contamination [79]. Additional limitations include susceptibility to transfer errors during serial incubation, data entry errors during manual recording, and operational inefficiencies in providing interim counts for manufacturing decisions [79].
These methodological constraints have direct consequences for pharmaceutical quality control. By the time a positive result is obtained using traditional methods—which can be anywhere from a few days to over two weeks—the opportunity to respond to the contamination event has long passed [76]. The impact can be substantial, resulting in product holds, line shutdowns, or even batch rejections that could potentially have been avoided with more timely detection.
Financial Analysis Frameworks for RMM Justification
Quantifying Return on Investment (ROI)
A rigorous financial analysis is essential for justifying RMM implementation. The Return on Investment (ROI) represents the ratio of money gained or lost on an investment relative to the amount invested. For RMM evaluation, ROI compares the cost of performing the conventional method with the costs and savings of implementing the new method. The standard formula for this calculation is:
ROI = [(Cost of CM - Cost of RMM) / Investment in RMM] × 100
Where CM represents the conventional method and RMM represents the rapid micro method [77]. Case studies demonstrate that robust ROI analysis can yield compelling financial justifications. One comprehensive evaluation of three different-sized manufacturing facilities found that implementing an RMM for environmental monitoring delivered first-year ROI ranging from 183% to 365%, with payback periods of just 3.3 to 6.6 months [76]. These figures illustrate that while initial investment is substantial, the operational savings can generate returns quickly and significantly.
Table 2: Financial Metrics for RMM Implementation in Three Facility Sizes
| Financial Metric | Small Facility | Medium Facility | Large Facility | Calculation Basis |
|---|---|---|---|---|
| Annual Active Air Samples | 40,000 | 70,000 | 100,000 | Environmental monitoring volume |
| Annual Product Lot Rejections Prevented | 1 | 1 | 3 | Contamination control improvement |
| Value of Rejected Lots | $300,000 | $1,000,000 | $1,500,000 | Batch value × rejections prevented |
| ROI (Year 1) | 183% | 255% | 365% | (Savings - Investment) / Investment |
| Payback Period | 6.6 months | 4.7 months | 3.3 months | Investment / Monthly Savings |
Comprehensive Cost-Benefit Analysis
Beyond simple ROI calculations, a comprehensive cost-benefit analysis for RMM implementation should incorporate multiple financial metrics that provide different perspectives on investment value. The Payback Period (PP) represents the time required for the return on investment to "repay" the sum of the original investment, calculated as the inverse of the ROI formula: PP = Investment in RMM / (Cost of CM - Cost of RMM) [77]. Net Present Value (NPV) provides insight into how much value an investment adds to the company over time, calculated by discounting future cash flows to their present value [77]. A positive NPV indicates that the RMM investment would add value to the organization, while a negative NPV suggests the investment would subtract value.
The business benefits contributing to these financial metrics include reduced testing and product release cycle times, reduction or elimination of laboratory equipment and overhead, lower headcount requirements, and decreased repeat testing and investigations [76]. Perhaps most significantly, RMMs can substantially reduce lot rejection, reprocessing, and rework because environmental monitoring excursions can be detected and addressed more quickly, preventing widespread contamination [76]. These cost avoidances often represent the most substantial financial benefit of RMM implementation, particularly in facilities manufacturing high-value pharmaceutical products.
Implementation Protocols and Validation Requirements
Experimental Validation Framework
The validation of RMMs requires a structured experimental approach to demonstrate equivalence or superiority to traditional methods. Regulatory guidelines, including those from USP Chapter <1223>, Ph. Eur. 5.1.6, and FDA CBER, provide frameworks for validation that typically require a combination of the following performance studies [7]:
- Accuracy: Demonstrating that the RMM produces results that correlate well with reference methods
- Precision: Determining the repeatability and reproducibility of the method under defined conditions
- Specificity: Confirming the method's ability to detect the target microorganisms in the presence of other microbial flora
- Limit of Detection: Establishing the lowest number of microorganisms that can be reliably detected
- Robustness: Evaluating the method's resilience to small, deliberate variations in method parameters
- Equivalence Testing: Statistical comparison with the conventional method using parallel testing [81]
A 2020 study exemplifies this validation approach, conducting parallel analysis using gold standard methods prescribed under U.S. Pharmacopeia alongside 3M Petrifilm RMMs [15]. The study tested five common assays—Total Aerobic Microbial Count, Total Yeast and Mold Count, Escherichia coli, Staphylococcus aureus, and Coliforms—across multiple dietary supplement categories, with three individual lots of each product tested to increase data robustness [15]. The acceptance criterion was set at demonstrating greater than 70% recovery compared to control, which was successfully achieved across all product categories with recovery ranges between 74-123% [15].
Implementation Workflow
Diagram: RMM Implementation Pathway
The implementation pathway for RMMs follows a logical sequence from assessment through validation and operational integration. The process begins with a comprehensive review of conventional methods to identify pain points and opportunities where RMMs could deliver significant improvements [76]. This assessment should quantify the costs associated with current methods, including labor, materials, and the financial impact of delays or investigations. The next phase involves technology evaluation and selection, where available RMM platforms are assessed against technical requirements and operational constraints [7]. This leads to business case development incorporating ROI analysis and other financial metrics to secure organizational buy-in and funding [76].
Following approval, the validation phase begins with detailed planning of the experimental protocols needed to demonstrate method suitability for its intended use [81]. The execution of validation protocols generates the data required to support both internal quality systems and regulatory submissions [7]. After successful validation, the method moves to regulatory submission (if required) and full implementation across the organization, followed by ongoing performance monitoring to ensure continued reliability and identify opportunities for further optimization [82].
Essential Research Tools and Reagent Solutions
Key Research Reagent Solutions
Table 3: Essential Research Reagents and Materials for RMM Implementation
| Reagent/Material | Function | Application Examples |
|---|---|---|
| PCR Reagents & Kits | Amplification and detection of microbial DNA/RNA | Sterility testing, pathogen detection in raw materials |
| Viability Markers & Dyes | Differentiation between live and dead cells | Flow cytometry, fluorescence-based assays |
| Selective Culture Media | Growth promotion of specific microorganisms | Method equivalence testing, validation studies |
| Reference Microbial Strains | System qualification and validation | Accuracy, precision, and robustness studies |
| Nucleic Acid Extraction Kits | Isolation of genetic material from samples | PCR-based RMM platforms |
| ATP Bioluminescence Reagents | Detection of microbial contamination via ATP measurement | Cleanroom monitoring, hygiene assessment |
The pharmaceutical rapid microbiology testing market reflects growing adoption, with the reagents and kits segment dominating product offerings due to their recurring use across various assays [78]. Major suppliers including Thermo Fisher Scientific, Merck KGaA, bioMérieux, and Danaher provide comprehensive reagent systems specifically validated for use with their RMM platforms [78]. These specialized reagents represent an ongoing operational expense that must be factored into the total cost of ownership for RMM systems, though they often replace the culture media and consumables required for traditional methods.
Market Context and Future Outlook
The global pharmaceutical rapid microbiology testing market is projected to grow from USD 1.25 billion in 2024 to approximately USD 6.29 billion by 2034, representing a robust compound annual growth rate of 17.54% [78]. This expansion is fueled by several converging trends, including increased adoption of nucleic acid-based technologies, integration of automation and robotics, and growing demand for real-time environmental monitoring in sterile manufacturing facilities [78]. The expansion of biologics and personalized medicine manufacturing represents a particularly significant opportunity, as these advanced therapies have short shelf lives and complex manufacturing processes that are incompatible with traditional microbiological methods requiring extended incubation periods [78].
Regulatory agencies have increasingly supported RMM implementation through initiatives like the FDA's Process Analytical Technology (PAT) framework and the establishment of consortia such as the NIST Rapid Microbial Testing Methods Consortium, which works to develop reference materials and validation frameworks [82]. This regulatory alignment, combined with continuous technological advancement, suggests that the financial justification for RMM implementation will become increasingly compelling as technologies mature and costs decrease.
Integrating Rapid Methods into Existing GMP Workflows and Quality Systems
Within pharmaceutical quality control and drug development, a significant transformation is underway, moving from traditional culture-based methods toward Rapid Microbiological Methods (RMM). This evolution is driven by the need for faster results, enhanced accuracy, and improved process control in Good Manufacturing Practice (GMP) environments. Traditional microbiological testing, while well-established and regulatory-recognized, often requires 48 to 72 hours or longer for incubation, creating bottlenecks in product release and process monitoring [1]. In contrast, RMMs can deliver results in hours or a few days, enabling real-time decision-making and significantly reducing the risk of contamination going undetected [83].
This guide objectively compares the performance of traditional and rapid methods, providing experimental data and protocols to support scientists and drug development professionals in evaluating and integrating these advanced technologies into existing quality systems. The shift towards a risk-based "Quality by Design" approach and initiatives like Process Analytical Technology (PAT) are creating a favourable regulatory climate for RMM adoption, as they provide the timely data essential for continuous process monitoring and parametric release [72].
Performance Comparison: Traditional vs. Rapid Microbial Methods
Quantitative Performance Data Analysis
The following table summarizes key performance metrics for various rapid methods compared to their traditional counterparts, based on published studies and manufacturer data.
Table 1: Performance Comparison of Traditional vs. Rapid Microbiological Methods
| Method Type | Specific Technology | Time-to-Result | Key Performance Metrics | Typical Applications |
|---|---|---|---|---|
| Traditional Plate Count | Colony Enumeration | 2-7 days (up to 14 for sterility) | Detects wide range of microbes; Standard for regulatory compendia [1]. | Bioburden, Environmental Monitoring, Sterility Testing [84] |
| Growth-based RMM | ATP Bioluminescence | 24-48 hours (with enrichment) | Measures cellular ATP; requires steps to reduce non-microbial ATP [72]. | Raw Material Testing, Water QC, Surface Monitoring [72] |
| Growth-based RMM | Autofluorescence (e.g., Growth Direct) | ~50% reduction vs. traditional | Detects microcolonies before visible to naked eye; non-destructive [72]. | Bioburden, Environmental Monitoring (air & surfaces) [72] |
| Viability-based RMM | Flow Cytometry | 1.5 - 2 hours | Detection limit of ~100 CFU/mL; uses fluorescent labeling [83]. | In-process testing, Water for Injection (WFI) quality control |
| Molecular Method | PCR (e.g., microproof) | A few hours | High specificity for targeted pathogens; requires microbial enrichment for low bioburden [72]. | Hygiene Screening, Specific Pathogen Detection [72] |
| Endotoxin Testing | LAL-based (e.g., Endosafe-PTS) | ~15 minutes | Quantitative, chromogenic assay; semi-automated cartridge system [72]. | Endotoxin testing for parenteral products and medical devices |
Key Comparative Studies and Experimental Data
Validation of a Rapid Method for Yeast and Mold
A 2023 study provides robust experimental data comparing the Soleris automated method against the traditional plate-count method for quantifying yeast and mold in an antacid oral suspension.
- Experimental Protocol: Testing was performed at three different microbial bioburden levels. Equivalence between the detection time of the Soleris method and the colony-forming units (CFUs) of the reference method was established using probability of detection, linear Poisson regression, Fisher's test, and multifactorial analysis of variance (ANOVA) [85].
- Results: All results from the rapid method were in statistical agreement with the reference plating procedures. The limits of detection and quantification were statistically similar for both methods (Fisher's exact test, P > 0.05). The method met all validation criteria, including precision (standard deviation <5, coefficient of variance <35%), accuracy (>70%), and linearity (R2 >0.9025) [85].
Rapid Identification in Clinical Blood Cultures
A 2020 study demonstrated the efficacy of a rapid centrifugation and Gram staining method for positive blood cultures, highlighting principles applicable to pharmaceutical in-process controls.
- Experimental Protocol: From 152 blood culture bottles showing positive growth signals within 12 hours, 5 mL of fluid was centrifuged. The resulting bacterial film layer was used for Gram staining, followed by identification and antibiotic susceptibility testing using an automated system [5].
- Results: The rapid method demonstrated 92% agreement with routine procedures for bacterial strain identification. For antibiotic susceptibility, a 97.4% agreement was observed across 1,984 comparative tests, enabling clinicians to report results with a high success rate (~97%) within 24 hours [5].
Essential Research Reagent Solutions for RMM Implementation
Successful integration of RMMs relies on specific reagents and materials. The following table details key solutions required for method validation and routine use.
Table 2: Key Research Reagent Solutions for Rapid Microbiological Methods
| Reagent/Material | Function | Application Example |
|---|---|---|
| Quality Control (QC) Organisms | Well-characterized microorganisms with defined profiles used to validate testing methodologies, monitor instrument/reagent performance, and conduct growth promotion testing of media [86]. | BIOBALL Custom Services for preserving in-house isolates; Microbiologics microbial controls for routine QC [86]. |
| Validated Culture Media | Supports microbial growth for both traditional and growth-based RMMs; requires growth promotion testing with QC strains to ensure performance [86]. | Used in autofluorescence systems (e.g., Growth Direct) where membranes are incubated on conventional nutrient media [72]. |
| Specialized Substrates & Enzymes | Enable specific detection technologies. Luciferin/luciferase for ATP bioluminescence; specific substrates for colorimetric growth detection [72]. | ATP-bioluminescence systems for assessing contamination in raw materials and filterable samples [72]. |
| Nucleic Acid Reagents | For molecular RMMs like PCR. Include primers, probes (e.g., TaqMan), and master mixes for amplifying and detecting target microbial DNA/RNA [84]. | Microsart qPCR kits for rapid sterility testing and mycoplasma detection [84]. |
| Reference Standards & Controls | Certified reference materials (CRMs) and proficiency test standards used for method calibration, ensuring data accuracy, and supporting lab accreditation [86]. | Zeptometrix ISO-accredited CRMs and proficiency test standards for ensuring data accuracy [86]. |
Implementation Roadmap: Integrating RMM into GMP Workflows
Regulatory and Validation Framework
Integrating a new RMM into a validated GMP workflow requires careful planning and adherence to regulatory guidance. Key steps include:
- Method Validation: Demonstration that the RMM is equivalent or superior to the conventional method it replaces. This is guided by PDA Technical Report 33, USP <1223>, and Ph. Eur. 5.1.6 [72] [87]. Critical validation parameters include accuracy, precision, specificity, limit of detection, and robustness [85].
- Regulatory Strategy: In the U.S., the FDA encourages discussion via the PAT initiative and allows for submission strategies like the Comparability Protocol (CP), a pre-approved validation plan that simplifies the implementation process for multiple products [87]. In the EU, the Post Approval Change Management Protocol (PACMP) serves a similar function as a two-step Type II Variation [87].
Workflow Integration and Automation
Automated RMMs eliminate unnecessary hands-on labor and manual data entry, increasing the productivity of skilled technicians while reducing human-based errors [88]. This is particularly valuable for applications like Environmental Monitoring, Bioburden, and Water Testing [88]. The diagram below illustrates the logical pathway for integrating a rapid method into an established GMP quality system.
The integration of Rapid Microbiological Methods into existing GMP workflows represents a significant advancement in pharmaceutical quality control. While traditional methods remain the regulatory gold standard for many applications, the compelling benefits of RMMs—dramatically reduced time-to-result, enhanced accuracy, automation, and improved data integrity—are driving their adoption [88] [1] [83]. The successful implementation of an RMM requires a strategic approach that includes robust technical and business due diligence, rigorous validation against compendial methods, and a clear regulatory strategy. By leveraging the frameworks and data presented in this guide, researchers, scientists, and drug development professionals can make informed decisions to modernize their microbiological quality systems, ultimately enhancing patient safety and manufacturing efficiency.
Validating for Compliance: ISO 16140, USP <1223>, and Equivalence Testing
In the field of pharmaceutical contamination detection, a significant transition is underway, moving from traditional, culture-based methods toward rapid microbiological methods (RMMs). This shift is driven by the need for faster results, enhanced sensitivity, and improved process control in drug development and manufacturing. Regulatory standards from the United States Pharmacopeia (USP), the European Pharmacopoeia (Ph. Eur.), and the International Organization for Standardization (ISO) provide the critical framework governing both traditional and innovative approaches. For decades, traditional microbiological techniques, such as the plate count method, have served as the established standard for most industries. These methods involve incubating samples on a petri dish for 48 to 72 hours, after which colony-forming units (CFUs) are counted to estimate microbial load [1]. While well-accepted by regulatory agencies like the FDA and proven in their accuracy and reliability, their primary drawback is the extended time-to-result, which can delay product release and process adjustments [1] [22].
The emergence of Rapid Microbiological Methods (RMMs) addresses this limitation by leveraging advanced technologies to significantly reduce detection time. RMMs can provide results in hours or even minutes, enabling quicker decision-making and enhanced contamination control [1] [72]. The regulatory landscape has evolved to accommodate these technological advancements. The Pharmacopeial Discussion Group (PDG), comprising USP, Ph. Eur., and the Japanese Pharmacopoeia (JP), works toward international harmonization of excipients and general methods, though differences still exist for a large number of general methods [89]. Furthermore, regulatory bodies now provide specific chapters, such as USP <1223> and Ph. Eur. 5.1.6, which outline the validation requirements for these alternative methods, ensuring they are accurate, reliable, and comparable to compendial methods [90]. This guide provides an objective comparison of traditional and rapid microbiological methods, framed within the context of regulatory standards from ISO, USP, and Ph. Eur., to aid researchers and drug development professionals in navigating this evolving field.
Understanding the Regulatory Landscape
Navigating the complex web of international pharmacopeias is fundamental for pharmaceutical development and global market access. The USP and Ph. Eur. are among the most influential pharmacopeias worldwide, with compliance being legally mandated in their respective jurisdictions [91] [89]. The US FDA enforces USP monographs, while Ph. Eur. is mandatory in 39 member states and applied in over 100 countries [89]. The Pharmacopeial Discussion Group (PDG) actively works to harmonize excipient monographs and general chapters among USP, Ph. Eur., and JP to streamline international compliance [91] [92]. Despite these efforts, many general methods have not yet been harmonized, requiring companies to develop multi-compendial testing strategies [89].
A key development in the regulatory approach is the increased emphasis on risk-based practices and quality by design, championed by initiatives like the FDA's Process Analytical Technology (PAT) [72]. PAT is defined as "a system for designing and controlling manufacturing through timely measurements of critical quality and performance attributes" with the goal of ensuring final product quality [72]. This framework creates a favorable environment for RMMs, as traditional culture-based methods are of limited value in PAT due to their long incubation times [72]. The regulatory path for implementing an RMM is clearly defined. USP General Chapter <1223> "Validation of Alternative Microbiological Methods" and Ph. Eur. Chapter 5.1.6 "Alternative Methods for Control of Microbiological Quality" provide the structured frameworks for demonstrating that a rapid method is fit-for-purpose and equivalent to the compendial method [90]. The Parenteral Drug Association's Technical Report 33 (TR-33) also offers detailed validation guidance [72] [7]. Successful validation and regulatory acceptance of several rapid product release test methods by the FDA demonstrate that a pathway for implementation exists, particularly for novel therapies like cell and gene products with short shelf lives [72].
Comparative Analysis: Traditional vs. Rapid Microbial Methods
Traditional and rapid microbiological methods differ fundamentally in their approach to detecting, enumerating, and identifying microorganisms. The following table provides a structured comparison of their core characteristics, supported by quantitative data where available.
Table 1: Comparative Analysis of Traditional and Rapid Microbiological Methods
| Feature | Traditional Methods | Rapid Microbiological Methods (RMMs) |
|---|---|---|
| Core Principle | Culture-based growth on solid or liquid media to form visible colonies [1] [22] | Detection of microbial biomarkers, metabolic activity, or nucleic acids without relying on visible growth [1] [72] |
| Time to Result | 48-72 hours for bioburden; up to 14 days for sterility tests [1] [22] | Minutes to hours; typically at least 50% faster than traditional methods [1] [72] [7] |
| Primary Output | Colony forming units (CFUs) [1] | Relative light units (ATP), fluorescence, nucleic acid sequences, etc. [72] [7] |
| Sensitivity | Can detect a wide range of microbes but may miss slow-growers or VBNC states [1] [7] | High sensitivity; capable of detecting low levels of contamination and VBNC states [1] [72] |
| Automation Level | Low; labor-intensive processes [1] | High; many systems are fully automated, reducing human error [1] [72] |
| Regulatory Status | Well-established and widely accepted gold standard [1] [7] | Growing acceptance; requires rigorous validation per USP <1223> and Ph. Eur. 5.1.6 [72] [90] |
| Initial Investment | Low equipment requirements [1] | High initial investment for instrumentation [1] [7] |
Categorization of Rapid Method Technologies
RMMs encompass a diverse range of technologies, which can be broadly categorized based on their underlying detection principle. The following table details the main categories, their technologies, and applications.
Table 2: Categories of Rapid Microbiological Methods and Their Applications
| Category | Technology Examples | How It Works | Typical Time to Result | Common Pharmaceutical Applications |
|---|---|---|---|---|
| Growth-Based | ATP Bioluminescence, Colorimetric Growth Detection, Autofluorescence [72] | Detects biochemical or physiological indicators of growth (e.g., ATP, CO₂ production) rather than visible colonies [72] | 24-48 hours (may include enrichment) [72] | Bioburden assessment, raw material testing, water quality control [72] |
| Viability-Based | Flow Cytometry, Solid-Phase Cytometry [72] [7] | Uses cell labeling with fluorescent stains to detect and quantify viable microorganisms [72] | Minutes to a few hours [72] [7] | Rapid sterility testing, microbial enumeration in filters [72] |
| Molecular Methods | Polymerase Chain Reaction (PCR), Real-Time PCR [1] [72] | Amplifies and detects specific sequences of microbial nucleic acids (DNA/RNA) [1] [72] | A few hours [1] [72] | Specific pathogen detection, identification of contaminants from sterility failures [72] [7] |
| Endpoint & Toxin Tests | Limulus Amebocyte Lysate (LAL) Assay [72] | Detects endotoxins from Gram-negative bacteria using a chromogenic or turbidimetric assay [72] | ~15 minutes [72] | Endotoxin testing for injectable pharmaceuticals [72] |
Experimental Protocols and Validation Frameworks
Validation of Rapid Microbiological Methods
The successful implementation of any RMM in a regulated environment hinges on a rigorous validation process that demonstrates the method is fit for its intended purpose. The core guidance documents for this process are USP <1223> and Ph. Eur. 5.1.6 [90]. The overarching goal of validation is to provide documented evidence that the RMM is at least equivalent to the traditional compendial method in terms of accuracy, reliability, and robustness [7] [90]. The validation process is a multi-stage endeavor, as outlined in the workflow below.
RMM Validation Workflow
Detailed Validation Parameters and Experimental Protocols
The workflow above is operationalized through the experimental assessment of specific validation parameters. The following details the experimental approach for each, as per regulatory guidelines [90]:
- Accuracy: This measures the closeness of agreement between the value found by the RMM and a reference value. The experiment involves testing samples spiked with known concentrations and types of microorganisms (e.g., E. coli, S. aureus, P. aeruginosa, C. albicans) and comparing the recovery rate to that of the compendial method (e.g., plate count) [90].
- Precision: This evaluates the reproducibility of the method under defined conditions. It is assessed at two levels:
- Repeatability: The same analyst tests homogeneous samples with the same equipment in a short interval.
- Intermediate Precision: Different analysts on different days using different instruments test the same homogeneous samples. Results are expressed as the standard deviation or relative standard deviation [90].
- Specificity: This demonstrates the method's ability to detect the target organism(s) unequivocally in the presence of other components, such as the product matrix or normal flora. Experiments involve testing the product matrix without microbes (to check for interference), the matrix spiked with target organisms, and the matrix spiked with related but non-target organisms [90].
- Limit of Detection (LOD) and Quantification (LOQ): The LOD is the lowest number of microorganisms the method can reliably detect. The LOQ is the lowest number that can be quantified with acceptable precision and accuracy. These are determined by progressively diluting a microbial suspension and analyzing it with the RMM [90].
- Robustness: This parameter assesses the method's capacity to remain unaffected by small, deliberate variations in method parameters (e.g., temperature, incubation time, reagent lot, operator). It demonstrates the reliability of the method during normal usage [90].
A critical step in validation is matrix interference testing. Product formulations (e.g., gels, creams, high-sugar solutions) can inhibit microbial detection or cause false positives. Validation must include studies where the product is spiked with known, low levels of microorganisms to demonstrate that the matrix does not interfere with recovery rates compared to the compendial method [90].
The Scientist's Toolkit: Key Reagents and Materials
Successful execution of both traditional and rapid microbiological tests requires specific reagents and materials. The following table lists essential items and their functions in the context of contamination detection research.
Table 3: Essential Research Reagents and Materials for Microbial Testing
| Item | Function/Description | Typical Application |
|---|---|---|
| Culture Media | Nutrient-rich substances (e.g., Tryptic Soy Agar, Fluid Thioglycollate Medium) designed to support microbial growth [22]. | Used in traditional methods for microbial enumeration, sterility testing, and growth promotion testing [22]. |
| ATP Bioluminescence Reagents | A specific combination of the substrate luciferin and the enzyme luciferase. The reaction with microbial ATP produces visible light, measured in relative light units (RLUs) [72]. | Core component of ATP-based RMMs for rapid hygiene monitoring and bioburden estimation after a short enrichment [72]. |
| PCR Master Mix | A pre-mixed solution containing reagents essential for PCR, including DNA polymerase, dNTPs, primers, and buffers. Primers target conserved microbial genetic regions (e.g., 16S rRNA) [72] [7]. | Used in molecular-based RMMs for the highly specific detection and identification of microorganisms; enables differentiation at the species level [72] [7]. |
| LAL Reagent | Limulus Amebocyte Lysate, derived from horseshoe crab blood. It contains enzymes that clot in the presence of bacterial endotoxins [72]. | The gold standard for endotoxin testing in injectable pharmaceuticals; used in rapid, cartridge-based systems for near real-time results [72]. |
| Sterile Diluents with Surfactants | Aqueous solutions (e.g., Buffered Sodium Chloride-Peptone Solution) often supplemented with surfactants like lecithin or polysorbate 80 [22]. | Used for sample homogenization and dilution; surfactants aid in neutralizing antimicrobial properties and achieving a homogenous microbial suspension [22]. |
| Reference Standards | Well-characterized microbial strains obtained from national culture collections (e.g., ATCC) [89]. | Critical for method validation, equipment qualification, and ensuring the accuracy and reproducibility of both traditional and rapid tests [89]. |
The journey from traditional, culture-based methods to rapid microbiological methods represents a significant evolution in pharmaceutical quality control. While traditional methods remain the legally recognized standard for many products and are valued for their simplicity and low initial cost, their lengthy incubation periods are a major bottleneck in an era that demands real-time process understanding and faster product release [1] [22]. RMMs, enabled by technologies such as ATP bioluminescence, flow cytometry, and PCR, offer a compelling alternative through dramatically reduced time-to-result, increased automation, and often superior sensitivity for detecting viable but non-culturable organisms [1] [72] [7].
The regulatory roadmap for implementing RMMs is clearly defined by USP <1223> and Ph. Eur. 5.1.6, which provide a robust framework for validation and demonstration of equivalence [90]. Although the initial investment and validation effort can be substantial, the long-term benefits—including faster release times, reduced inventory costs, improved contamination control, and enhanced patient safety—present a strong business and scientific case for adoption [72] [90]. As the regulatory climate continues to encourage innovation through PAT and quality by design initiatives, and as novel therapies with short shelf-lives become more common, the adoption of rapid microbiological methods is poised to accelerate, solidifying their role as the future of microbiology in pharmaceutical research and manufacturing.
In pharmaceutical microbiology, the reliability of contamination detection methods is paramount for ensuring drug safety and quality. Validation parameters provide the foundational framework for demonstrating that analytical methods are fit for their intended purpose, whether using traditional culture-based approaches or modern rapid microbiological methods (RMMs). These parameters—specificity, limit of detection (LOD), limit of quantitation (LOQ), and robustness—form the critical benchmarks for evaluating any method's performance [93] [22]. As the pharmaceutical industry increasingly adopts RMMs to overcome the time constraints of traditional techniques, understanding these validation parameters becomes essential for researchers, scientists, and drug development professionals [1] [8]. This guide objectively compares how these core validation parameters apply across methodological approaches, supported by experimental data and implementation protocols.
Core Validation Parameters Explained
Validation parameters ensure that analytical methods consistently produce reliable, accurate, and meaningful results. The International Council for Harmonisation (ICH) guidelines Q2(R2) and Q14 provide the primary framework for these parameters in pharmaceutical applications [93].
Specificity: The parameter of method selectivity that ensures the ability to unequivocally assess the analyte in the presence of other components. For microbiological methods, this translates to accurately detecting or quantifying target microorganisms without interference from product components, media, or other microorganisms [93]. Specificity is particularly challenging for RMMs that rely on surrogate markers like ATP bioluminescence, which may detect non-microbial sources, or PCR methods that must distinguish between viable and non-viable cells [1] [7].
Limit of Detection (LOD): The lowest concentration of microorganisms that can be detected, but not necessarily quantified, under stated experimental conditions. This parameter is crucial for sterility testing and contamination screening where even single cells must be detected [93] [22]. Rapid methods often demonstrate superior LOD compared to traditional approaches, with some technologies capable of detecting viable but non-culturable (VBNC) organisms that traditional methods miss [7] [8].
Limit of Quantitation (LOQ): The lowest number of microorganisms that can be quantitatively determined with acceptable precision and accuracy. This parameter is essential for microbial enumeration tests where the exact concentration must be measured, such as in bio-burden testing [93]. While traditional methods rely on colony-forming unit (CFU) counts, rapid methods use alternative quantification metrics like relative light units (RLU) in ATP bioluminescence or genomic copies in PCR, creating challenges in comparative analysis [8].
Robustness: The measure of a method's reliability and consistency during normal usage, demonstrated by its capacity to remain unaffected by small, deliberate variations in method parameters. It provides an indication of the method's reliability during normal use and is closely linked to ruggedness, which addresses interlaboratory variation [93] [94]. Robustness is particularly important for methods deployed across multiple laboratories with different operators, equipment, and environmental conditions [94].
Table 1: Validation Parameter Definitions and Regulatory Significance
| Parameter | Technical Definition | Importance in Microbiological Testing | Regulatory Reference |
|---|---|---|---|
| Specificity | Ability to distinguish target microorganisms from interfering components | Ensures detection accuracy in complex product matrices | ICH Q2(R2) [93] |
| LOD | Lowest number of detectable microorganisms | Critical for sterility testing and contamination control | FDA Analytical Procedures [93] |
| LOQ | Lowest number of quantifiable microorganisms with precision and accuracy | Essential for microbial enumeration and bio-burden testing | ICH Q2(R2) [93] |
| Robustness | Method performance under deliberate parameter variations | Predicts consistent performance in different laboratories | ICH Q14 [93] |
Comparative Analysis: Traditional vs. Rapid Methods
Performance Comparison Across Validation Parameters
Traditional microbiological methods, including colony counting and most probable number (MPN) techniques, have served as gold standards for decades. These methods are well-established, widely accepted by regulatory bodies, and provide proven accuracy for detecting a wide range of microorganisms [1]. However, they suffer from being time-consuming (24-72 hours or longer) and labor-intensive, with limitations in sensitivity particularly for slow-growing or VBNC organisms [1] [22].
Rapid microbiological methods encompass diverse technologies including nucleic acid-based detection (PCR), enzyme-based methods (ATP bioluminescence), and optical systems (flow cytometry) [8]. These methods offer significantly faster results (hours rather than days), higher sensitivity, and greater potential for automation [1] [7]. However, they face challenges including high initial investment costs, limited applicability across all sample types, and ongoing regulatory adaptation [1] [8].
Table 2: Method Comparison Across Validation Parameters
| Validation Parameter | Traditional Methods | Rapid Microbial Methods |
|---|---|---|
| Specificity | Detects viable microorganisms through growth; may miss VBNC states [8] | Technology-dependent; PCR highly specific; ATP may have interference [7] |
| LOD | ~1 CFU detectable after prolonged incubation (48-72 hours) [22] | Often lower; can detect single cells in hours; can detect VBNC [8] |
| LOQ | Manual CFU counting; subjective with operator variability [22] | Automated quantification; objective but may require correlation to CFU [8] |
| Robustness | Well-documented through decades of use; media and incubation variations studied [1] | Emerging data; equipment and reagent stability considerations [7] |
| Regulatory Status | Fully established in compendial standards [1] | Increasing acceptance; case-by-case validation required [7] [8] |
Experimental Data and Case Studies
Quantitative comparisons between methods require careful experimental design. A robust approach involves building comparison pairs between candidate and comparative methods, selecting appropriate statistical analyses based on whether methods use the same detection principle, and setting predefined acceptance criteria [95].
For detection limit studies, research demonstrates that rapid methods generally provide equivalent or superior sensitivity compared to traditional approaches. One study found that ATP bioluminescence and PCR-based methods could detect contamination 24-48 hours faster than conventional culture methods, with some technologies achieving detection limits of 1-10 CFU/mL depending on the microorganism and technology used [8].
In robustness testing, a hierarchical experimental design examining multiple laboratories, analysts, and samples demonstrated that robust statistical methods provide more reliable variance component estimates than traditional ANOVA, particularly with the variable data typical of microbial enumeration [94]. This approach helps quantify the interlaboratory reproducibility critical for method transfer.
Experimental Protocols for Validation Studies
Specificity and Selectivity Testing
Protocol for Traditional Methods:
- Interference Testing: Prepare product samples without intentional contamination alongside inoculated samples. Use standard culture conditions (e.g., Soybean-Casein Digest Medium at 30-35°C for bacteria, 20-25°C for fungi) for 3-5 days [22].
- Challenge Microorganisms: Inoculate separate samples with representative compendial strains (e.g., E. coli, S. aureus, P. aeruginosa, S. cerevisiae, A. brasiliensis) at approximately 100 CFU per sample.
- Recovery Calculation: Compare CFU counts from challenge samples to positive controls without product matrix.
- Acceptance Criterion: Growth in challenge samples should not differ significantly from controls (typically ≥70% recovery) [22].
Protocol for Rapid Methods (PCR Example):
- Primer Specificity: Test in silico analysis against genomic databases followed by empirical testing against a panel of closely related non-target microorganisms.
- Matrix Interference: Spike target microorganisms into product matrix and compare detection to buffer controls.
- Inhibition Testing: Include internal amplification controls to detect PCR inhibitors in the sample matrix.
- Cross-Reactivity: Test against a panel of non-target microorganisms that might be present in the manufacturing environment [7].
LOD and LOQ Determination
Traditional Methods (MPN Approach):
- Prepare serial dilutions of target microorganisms with known concentrations.
- Inoculate multiple replicates (typically 3-5) at each dilution level into appropriate liquid media.
- Incubate under standard conditions and record growth/no growth for each replicate.
- Apply statistical MPN tables to determine the detection limit with 95% confidence intervals.
- For LOQ, use plate count methods with decreasing inoculum levels, defining LOQ as the lowest concentration with ≤15% CV between replicates [22].
Rapid Methods (Probabilistic Approach):
- Prepare samples at approximately 1-10 CFU per sample unit.
- Test a minimum of 20 replicates at this low concentration.
- Calculate LOD as the concentration where ≥95% of replicates test positive.
- For LOQ, test multiple replicates across a concentration range, determining where the coefficient of variation (CV) falls below 15% [93] [8].
Robustness and Ruggedness Testing
Protocol for Both Method Types:
- Experimental Design: Implement a nested design with multiple laboratories (≥3), analysts per laboratory (≥2), and samples per analyst (≥2) with replication [94].
- Deliberate Variations: For robustness, introduce small, intentional method parameter changes (e.g., incubation temperature ±2°C, incubation time ±10%, reagent lots, equipment).
- Statistical Analysis: Use robust statistical methods rather than traditional ANOVA to accommodate potential outliers in microbial data:
- Apply robust ANOVA using an M-estimator approach
- Alternatively, use Remedian (recursive median) method
- Calculate variance components for each experimental level [94]
- Acceptance Criteria: Method performance should remain within predefined specifications across all variations.
Quantitative Comparison Framework
The following workflow outlines a systematic approach for comparing traditional and rapid methods across key validation parameters:
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Research Materials for Validation Studies
| Category | Specific Items | Application in Validation Studies |
|---|---|---|
| Culture Media | Soybean-Casein Digest Medium, Tryptic Soy Agar, Sabouraud Dextrose Agar [22] | Supports microorganism growth in traditional methods; quality critical for performance |
| Reference Strains | ATCC/ NCTC strains (e.g., E. coli ATCC 8739, S. aureus ATCC 6538) [22] | Standardized microorganisms for specificity, LOD, and accuracy studies |
| Sample Preparation | Sterile diluents (buffered saline), surfactants (lecithin, polysorbate), neutralizing agents [22] | Extract and recover microorganisms from product matrices without inhibition |
| Molecular Biology Reagents | Primers/probes, polymerase enzymes, dNTPs, DNA extraction kits [7] | Essential for nucleic acid-based RMMs; reagent quality affects LOD and specificity |
| Bioluminescence Reagents | Luciferase enzyme, luciferin substrate, ATP standard solutions [8] | ATP-based detection systems; requires careful handling and standardization |
| Quality Controls | Positive controls, negative controls, internal amplification controls [93] | Monitor method performance during validation; essential for robustness assessment |
Implementation Roadmap and Regulatory Considerations
Successfully implementing microbiological methods requires careful planning and regulatory strategy. The following pathway outlines key stages from selection through ongoing monitoring:
Regulatory acceptance of RMMs continues to evolve, with guidelines from FDA, USP, Ph. Eur., and ICH providing frameworks for validation [93]. Successful regulatory strategy should include:
- Thorough comparative data against compendial methods
- Justification of method selection for intended purpose
- Complete validation data across all relevant parameters
- Risk assessment of new technology limitations
- Change control procedures for post-approval modifications [7] [93]
For traditional methods, the focus remains on demonstrating consistency with compendial standards, while RMMs require more extensive equivalence testing and scientific justification [8].
Validation parameters provide the critical framework for evaluating microbiological methods across the pharmaceutical industry. While traditional methods offer regulatory familiarity and established robustness, rapid methods provide clear advantages in speed, sensitivity, and objectivity. The choice between methodological approaches must balance regulatory compliance, operational efficiency, and scientific advancement.
As the industry continues its gradual transition toward rapid methods, understanding these validation fundamentals becomes increasingly important. By applying structured comparative approaches and robust statistical analyses, researchers can make informed decisions about method suitability while building the scientific evidence base needed for regulatory acceptance. The demystification of these validation parameters empowers scientists to advance contamination control strategies while maintaining the rigorous quality standards essential for pharmaceutical products.
The validation of rapid microbiological methods against traditional culture-based techniques is a critical process in pharmaceutical, food, and clinical laboratories. Demonstrating method equivalence ensures that new technologies provide reliable, accurate, and reproducible results while offering advantages in speed, sensitivity, or automation [22] [96]. This guide outlines the structured protocols for method comparison and interlaboratory studies, which are essential for regulatory acceptance and implementation of alternative methods.
Traditional microbiological methods are often labor-intensive and time-consuming, requiring incubation periods ranging from 2 to 7 days, and up to 14 days for sterility testing of pharmaceutical formulations [22]. Rapid methods, including polymerase chain reaction (PCR), enzyme-linked immunosorbent assay (ELISA), ATP bioluminescence, impedance microbiology, and biosensors, offer significantly faster results with improved sensitivity and precision [22] [97] [98]. However, before these methods can be adopted, a rigorous demonstration of equivalence must be completed following established standards and regulatory frameworks.
Regulatory Frameworks and Standards
Key Regulatory Guidelines
Several international standards provide frameworks for validating alternative microbiological methods. The United States Pharmacopeia (USP) Chapter <1223> outlines comprehensive guidance for validation of alternative microbiological methods used in pharmaceutical industries [96]. Similarly, the ISO 16140 series establishes protocols for method validation in the food chain, addressing both proprietary alternative methods and non-proprietary methods [99].
USP <1223> applies to alternative methods used for microbial enumeration, identification, detection, antimicrobial effectiveness testing, and sterility testing [96]. It requires demonstration of equivalent or better performance compared to compendial methods through defined validation criteria.
The ISO 16140 series consists of multiple parts covering vocabulary, protocol for validation of alternative methods against reference methods, verification in single laboratories, and factorial interlaboratory validation [99]. This standard is particularly important for food and feed testing laboratories, test kit manufacturers, and food business operators.
Validation Criteria Requirements
According to USP <1223>, validation of alternative methods must address several key performance aspects [96]:
- Accuracy: The closeness of agreement between the value found and the reference value
- Precision: The degree of agreement among individual test results
- Specificity: The ability to detect target microorganisms in the presence of related organisms
- Limit of Detection: The lowest number of microorganisms that can be detected
- Limit of Quantification: The lowest number of microorganisms that can be enumerated
- Robustness: The reliability of analysis under normal but variable operating conditions
- Linearity: The ability to produce results proportional to analyte concentration
Table 1: Validation Criteria Requirements for Alternative Microbiological Methods
| Validation Parameter | Qualitative Methods | Quantitative Methods | Key Considerations |
|---|---|---|---|
| Accuracy | Required | Required | Comparison to reference method results |
| Precision | Not required | Required | Repeatability and intermediate precision |
| Specificity | Required | Required | Includes inclusivity and exclusivity testing |
| Limit of Detection | Required | Not required | For detection methods only |
| Limit of Quantification | Not required | Required | For enumeration methods only |
| Linearity | Not required | Required | Across specified quantification range |
| Range | Not required | Required | Confirmation of accurate quantification |
| Robustness | Recommended | Recommended | Evaluation of method reliability |
Experimental Protocols for Method Comparison
Method Comparison Study Design
The initial stage of demonstrating equivalence involves a method comparison study, typically conducted by a single laboratory [99]. This study follows a structured protocol to compare the performance of the alternative method against the reference method.
Sample Panel Preparation: A representative panel of samples must be selected, including artificially contaminated samples and naturally contaminated samples when possible. For food testing, samples should represent different categories in the food chain, such as heat-processed milk and dairy products, meats, and ready-to-eat foods [99]. The sample size should provide sufficient statistical power, typically including multiple replicates across different contamination levels.
Blinded Testing: Both the alternative and reference methods should be applied to the same sample panels under blinded conditions to prevent bias. For qualitative methods, the comparison focuses on detection capability, while quantitative methods assess enumeration accuracy [99] [96].
Statistical Analysis for Equivalence
The data generated from method comparison studies require appropriate statistical analysis to demonstrate equivalence. For quantitative methods, regression analysis, correlation coefficients, and difference plots (such as Bland-Altman) are used to assess agreement between methods [96]. For qualitative methods, statistical measures include relative accuracy, relative detection level, probability of detection, and false positive/negative rates [99].
The alternative method should show non-inferiority compared to the reference method. Acceptance criteria must be predefined based on the method's intended use and regulatory requirements [96]. A comprehensive statistical analysis should demonstrate that the alternative method meets or exceeds these criteria.
Interlaboratory Study Protocols
Study Design and Organization
Interlaboratory studies represent the second validation stage, providing critical data on method performance across different laboratory environments, equipment, and personnel [99]. These studies are essential for demonstrating method robustness and transferability.
Participant Selection: A sufficient number of laboratories must participate to provide meaningful statistical data. The ISO 16140-2 standard specifies the required number of participants based on the validation type [99]. Participants should represent typical user laboratories with varying expertise levels.
Sample Distribution: Homogeneous and stable samples are distributed to all participating laboratories. Sample panels should include blank samples, artificially contaminated samples at various levels, and confirmed positive samples [99] [100]. The sample coding should be blinded to prevent bias during testing.
Protocol Standardization: All participating laboratories must follow the same standardized protocol for both the reference and alternative methods. This includes detailed instructions for sample preparation, method execution, and data reporting [99].
Data Analysis and Evaluation
The data collected from interlaboratory studies are analyzed to determine method performance characteristics across multiple laboratories:
For quantitative methods: The data analysis includes calculation of repeatability standard deviation, reproducibility standard deviation, and estimation of method accuracy [99]. The results are evaluated against predefined acceptance criteria for precision and accuracy.
For qualitative methods: The analysis focuses on false positive rates, false negative rates, relative sensitivity, relative specificity, and relative accuracy compared to the reference method [99]. The alternative method must demonstrate comparable or superior performance to the reference method.
Table 2: Key Performance Indicators in Interlaboratory Studies
| Performance Indicator | Quantitative Methods | Qualitative Methods | Acceptance Criteria |
|---|---|---|---|
| Repeatability | Within-laboratory variability | Within-laboratory concordance | CV% or percentage agreement within limits |
| Reproducibility | Between-laboratory variability | Between-laboratory concordance | CV% or percentage agreement within limits |
| Accuracy | Comparison to reference values | Detection capability vs. reference | Statistically equivalent or superior |
| Specificity | Freedom from interference | False positive rate | Meets predefined criteria |
| Sensitivity | Detection at low levels | False negative rate | Meets predefined criteria |
The study by Cruz et al. (2013) provides an exemplary model for interlaboratory comparison of PCR methods for diagnosing human leishmaniasis [100]. Their protocol included a quality control step and reduced variability among samples tested by each participant, involving four laboratories from different endemic regions across four continents.
Implementation and Verification Protocols
Laboratory Implementation Verification
Once a method has been validated through interlaboratory studies, individual laboratories must demonstrate their ability to perform the method correctly. The ISO 16140-3 standard outlines a two-stage verification process for user laboratories [99].
Implementation Verification: The laboratory demonstrates that it can properly perform the validated method by testing one of the same items evaluated in the validation study. This confirms that the laboratory can achieve results comparable to those obtained during the validation study [99].
Item Verification: The laboratory extends verification to challenging food items within its specific scope of testing. This demonstrates that the method performs satisfactorily for the specific sample types routinely tested by the laboratory [99].
Documentation and Quality Assurance
Proper documentation is essential throughout the validation and verification process. The validation report should comprehensively document all testing performed, including method parameters, equipment used, and detailed results [96]. This documentation must be thorough enough to support regulatory submissions and withstand audit scrutiny.
Ongoing monitoring and maintenance after initial validation are necessary to ensure continued method performance. This includes periodic system suitability testing, calibration, and preventive maintenance according to established quality assurance protocols [96].
Research Reagent Solutions and Materials
Successful execution of method comparison and interlaboratory studies requires specific research reagents and materials. The following table details essential solutions and their functions in equivalence studies.
Table 3: Essential Research Reagent Solutions for Equivalence Studies
| Reagent/Material | Function | Application Examples |
|---|---|---|
| Certified Reference Materials | Provides standardized benchmarks for comparison; validates method accuracy [101] [102] | Quantification accuracy assessment; method calibration |
| Culture Media | Supports growth of microorganisms for reference methods [22] [97] | Traditional plate count methods; reference method execution |
| Selective Agar | Isolates specific microorganisms from mixed cultures [99] | Confirmation procedures; specificity testing |
| Sterile Diluents | Dilutes samples to appropriate concentration; maintains microbial viability [22] | Sample preparation; homogenization |
| Surfactants | Aids in achieving homogeneous sample suspensions [22] | Sample preparation for hydrophobic matrices |
| Molecular Detection Reagents | Enables nucleic acid-based detection (primers, probes, enzymes) [100] [98] | PCR-based alternative methods; genetic identification |
| Immunoassay Components | Facilitates antigen-antibody detection (antibodies, substrates) [98] | ELISA-based methods; rapid pathogen detection |
Workflow Visualization
The following diagram illustrates the complete protocol for demonstrating method equivalence through comparison and interlaboratory studies:
Validated Method Implementation Pathway - This workflow outlines the sequential stages for demonstrating method equivalence, from initial validation to routine implementation.
The protocol for demonstrating equivalence through method comparison and interlaboratory studies provides a rigorous framework for validating rapid microbiological methods against traditional approaches. By following established standards such as USP <1223> and ISO 16140, laboratories can generate robust scientific evidence supporting method equivalence [99] [96].
The successful implementation of these protocols enables laboratories to adopt innovative technologies that offer significant advantages in speed, sensitivity, and automation while maintaining scientific rigor and regulatory compliance. As technology continues to advance, these validation protocols will remain essential for ensuring the reliability and accuracy of microbiological methods across pharmaceutical, food, and clinical sectors.
Laboratory method verification stands as a critical gatekeeper in microbiological testing, ensuring that methods perform as expected within a specific laboratory environment. For researchers and drug development professionals, the introduction of ISO 16140-3:2021 has provided a much-needed, internationally recognized standard for verifying validated microbiological methods. Prior to its publication, laboratories accredited to ISO 17025 lacked a standardized protocol for this essential process, leading to potential inconsistencies [103]. This standard offers a structured framework for laboratories to demonstrate that they can satisfactorily perform a validated method and that the method is fit for its intended purpose, particularly in the context of comparing traditional and rapid microbiological methodologies [104].
The verification process confirms that a method is correctly implemented and aligned with the specific food matrices or sample types analyzed in the user laboratory [103]. This is especially crucial when transitioning from traditional microbial methods, which often rely on growth-based detection like plate counts, to Rapid Microbiological Methods (RMMs) that may utilize technologies such as PCR, MALDI-TOF MS, and biosensors [1] [7]. The core principle is to build confidence in test results, whether for quality control in pharmaceutical manufacturing, environmental monitoring, or product safety testing.
Traditional vs. Rapid Microbiological Methods: A Comparative Framework
The choice between traditional and rapid methods involves a nuanced trade-off between time, cost, sensitivity, and regulatory acceptance. The table below summarizes the key characteristics of each approach.
Table 1: Comparison of Traditional and Rapid Microbiological Methods
| Characteristic | Traditional Methods | Rapid Methods (RMMs) |
|---|---|---|
| Basis of Detection | Culture-based growth on agar plates (e.g., CFU counting) [1] | Molecular, spectroscopic, or enzymatic (e.g., PCR, MALDI-TOF MS, ATP bioluminescence) [1] [7] |
| Time to Result | 48 to 72 hours, or longer for slow-growing organisms [1] [7] | As little as a few hours to minutes for some technologies [1] [7] |
| Primary Advantages | Well-established, widely accepted by regulators (FDA, EPA, USDA), proven accuracy, low equipment costs [1] | High speed, automation (reduced human error), high sensitivity and specificity, cost-effective at high volumes [1] |
| Primary Limitations | Time-consuming, labour-intensive, limited sensitivity for low-level contamination [1] | High initial investment, limited applicability for some samples, ongoing regulatory validation for some technologies [1] [7] |
| Typical Applications | Standard sterility testing, microbial limit tests, environmental monitoring [7] | High-throughput screening, raw material testing, hygiene monitoring, rapid pathogen detection [1] [105] |
Rapid methods represent a significant advancement but also introduce new challenges. For instance, they can detect Viable But Non-Culturable (VBNC) organisms and stressed environmental organisms that traditional methods might miss, forcing laboratories to re-euminate their contamination control strategies [7]. Furthermore, while regulatory authorities are increasingly accepting of RMMs, the validation pathway requires careful planning and execution to prove that the new method is at least equivalent to the traditional one, as outlined in guidelines like USP Chapter <1223> and FDA CBER Draft guidance [7].
Experimental Comparisons and Supporting Data
Case Study 1: Rapid Identification in Blood Cultures
A critical study compared a rapid method against the routine method for identifying microorganisms from 152 positive blood culture samples. The rapid method involved centrifuging samples that showed positive growth within 12 hours, performing Gram staining, and then conducting identification and antibiotic susceptibility testing [5].
Table 2: Performance Metrics of Rapid vs. Routine Blood Culture Processing
| Parameter | Rapid Centrifugation Method | Routine Processing Method | Agreement |
|---|---|---|---|
| Identification Agreement | Identification via automated system after centrifugation | Colony growth on agar followed by MALDI-TOF MS identification | 92% (138/150 samples) [5] |
| Antibiotic Susceptibility Agreement | AST performed directly from centrifuged sample | AST performed from isolated colonies | 97.4% (1,934/1,984 assays) [5] |
| Reportable Time to Clinician | Results reported within 24 hours of positive signal | Requires 24-hour incubation after positive signal before AST can be set up | Significantly faster, allowing for earlier treatment [5] |
Experimental Protocol:
- Sample Preparation: 5 ml of liquid was taken from blood culture vials (Bactec FX) showing a positive signal within 12 hours.
- Centrifugation: Samples were centrifuged at 2,000 rpm for 10 minutes, creating a bacterial film layer above the gel in the collection tube.
- Gram Staining: Bacteria from the film layer were smeared on a slide and subjected to standard Gram staining (crystal violet, Lugol's solution, alcohol decolorization, diluted fuchsin) [5].
- Identification & AST: Bacteria were picked from the gel, diluted to a 0.5–0.63 McFarland standard, and analyzed using an automated system (Phoenix 100) with appropriate ID/AST kits [5].
Case Study 2: Microbiological Assay vs. HPLC for Antibiotic Quantification
A comparative study of clarithromycin quantification in human plasma demonstrated the performance differences between a microbiological agar well diffusion bioassay and a selective High-Performance Liquid Chromatography (HPLC) method.
Table 3: Performance of Microbiological Bioassay vs. HPLC for Clarithromycin Quantification
| Performance Metric | Microbiological Bioassay | HPLC Method |
|---|---|---|
| Correlation with Spiked Samples | Concordant with HPLC (R² = 0.871, p < 0.001) [106] | Reference method for spiked samples [106] |
| Precision (Relative Standard Deviation) | 4.51% - 26.78% (inter-assay) [106] | 0.88% - 19.86% (improved precision) [106] |
| Accuracy (% of Nominal Value) | 78.52% - 131.19% (inter-assay) [106] | 99.27% - 103.42% (improved accuracy) [106] |
| Linearity Range | 250 - 3,000 ng/ml [106] | 62.5 - 3,000 ng/ml (wider range) [106] |
Experimental Protocol (Bioassay):
- Agar Well Diffusion: An agar plate was inoculated with Micrococcus luteus ATCC 9341.
- Sample Application: Wells were punched in the agar and filled with 100 µl of calibration or test plasma samples.
- Incubation and Reading: Plates were incubated at 35°C for 24 hours, and the diameter of the inhibition zone was measured [106].
The study concluded that while the bioassay showed concordance with HPLC for spiked samples, it was less precise and accurate. Furthermore, in samples from dosed volunteers, the bioassay measured total antimicrobial activity, including active metabolites, while HPLC measured only the parent drug, highlighting a key methodological difference [106].
The ISO 16140-3:2021 Verification Workflow
The verification process under ISO 16140-3 is a logical sequence of steps to ensure a laboratory is technically competent to perform a method before implementing it for routine testing. The following diagram illustrates the key stages of this workflow.
The Scientist's Toolkit: Key Reagents and Materials
Successful implementation and verification of microbiological methods, whether traditional or rapid, rely on a suite of essential research reagents and materials.
Table 4: Essential Research Reagent Solutions for Microbiological Testing
| Item | Function and Application |
|---|---|
| Culture Media (e.g., TSA, TSB) | Supports the growth and proliferation of microorganisms for traditional culture-based methods and serves as a base for many rapid methods [107]. |
| Neutralizing Buffers & Contact Plates | Used in environmental sampling (e.g., TSAWLPZS contact plates) to inactivate residual disinfectants on surfaces, allowing for accurate recovery of viable microorganisms [107]. |
| Identification Kits & Broths (e.g., ID Broth, AST Broth) | Liquid media used in automated identification and antibiotic susceptibility testing (AST) systems to standardize inoculum preparation [5]. |
| Reference Strains (e.g., ATCC strains) | Genetically well-characterized microorganisms used as positive controls and for method validation and verification to ensure accuracy and reproducibility [106]. |
| Molecular Assay Components (for PCR) | Includes primers, probes, and master mixes essential for nucleic acid-based RMMs, enabling specific detection and quantification of target pathogens [105] [7]. |
| MALDI-TOF MS Matrix & Standards | A chemical matrix (e.g., HCCA) that co-crystallizes with microbial proteins, enabling ionization and analysis for rapid microbial identification by mass spectrometry [5]. |
The laboratory verification process mandated by ISO 16140-3:2021 provides a critical and standardized framework for ensuring the reliability of microbiological testing methods. As the pharmaceutical and biotechnology industries continue to evolve with an increasing focus on biologics and personalized medicine, the demand for robust, sensitive, and timely contamination detection will only grow [105]. The comparative data clearly shows that while Rapid Microbiological Methods (RMMs) offer transformative benefits in speed and automation, traditional methods remain a trusted and often required standard. The ultimate choice of method is context-dependent, hinging on the specific application, regulatory requirements, and the balance between speed and initial investment. For researchers and drug development professionals, a thorough understanding and diligent application of method verification is the indispensable step that underpins all subsequent data, ensuring product safety, efficacy, and ultimately, patient safety.
The detection and identification of microbial contamination are critical in pharmaceutical research, clinical diagnostics, and drug development. For decades, traditional culture-based methods have served as the cornerstone of microbiological analysis, relying on the growth of microorganisms on specific media and subsequent phenotypic identification [80] [1]. While well-established, these methods are often laborious and time-consuming, requiring several days to yield results, which can delay critical decisions in both manufacturing and clinical settings [8] [80].
In response to these limitations, rapid microbiological methods (RMMs) have emerged as innovative paradigms that leverage advances in molecular biology, genomics, and proteomics [80]. These include nucleic acid-based detection methods such as polymerase chain reaction (PCR) and next-generation sequencing (NGS), mass spectrometry-based techniques like MALDI-TOF, and viability-based technologies using cell labeling [72] [80]. These methods significantly reduce the time to result, with some providing answers in hours rather than days, and offer enhanced sensitivity and specificity for microbial detection [72] [80].
This guide objectively compares the performance of traditional cultural methods with rapid microbiological methods, focusing on analytical concordance, discrepancies in detection capabilities, and clinical relevance. We present supporting experimental data from recent studies to provide researchers, scientists, and drug development professionals with a comprehensive evidence base for method selection and implementation.
Performance Metrics and Quantitative Data Comparison
Studies directly comparing traditional and rapid methods reveal significant differences in detection capabilities, turnaround times, and concordance rates. The following table summarizes key quantitative findings from recent comparative studies across various applications.
Table 1: Quantitative Comparison of Traditional Culture vs. Rapid Molecular Methods
| Metric | Traditional Culture Methods | Rapid Molecular Methods (PCR/NGS) | Application Context | Study Reference |
|---|---|---|---|---|
| Organism Identification Concordance | Reference Method | 97.4% | Pneumonia (BioFire PN Panel) | [108] |
| Turnaround Time | 2-5 days | ~1 hour (hands-on time <5 min) | Pneumonia (BioFire PN Panel) | [108] |
| Detection Richness | 17 different bacterial taxa | 338 different bacterial taxa | Chronic Wounds (16S rDNA) | [109] |
| Probability of Culture Growth | 4% at 104 copies/mL, 53% at 107 copies/mL | Semiquantitative PCR bins (104 to 107 copies/mL) | Pneumonia Pathogen Load | [108] |
| Detection of Resistance Markers | Reference Method (phenotypic) | 99% Concordance | Pneumonia (Antimicrobial Resistance Genes) | [108] |
| Method Equivalence | Standard Plate Count (Reference) | Validated for Yeast/Mold (Probability of Detection, P>0.05) | Antacid Oral Suspension | [110] |
The data demonstrates that while concordance for organism identification is high when an organism is detected by both methods, molecular methods identify a substantially greater diversity of microorganisms. The semiquantitative nature of some rapid methods also provides valuable information on microbial load, which correlates with the probability of growth in culture [108]. This is crucial for distinguishing colonization from true infection in clinical settings.
Detailed Experimental Protocols and Methodologies
To ensure the validity of comparative data, understanding the underlying experimental protocols for both traditional and rapid methods is essential. The following workflows and descriptions outline standard methodologies cited in the research.
Traditional Culture-Based Protocol
Traditional methods rely on microbial growth and phenotypic identification. The following diagram illustrates a general workflow for traditional bacterial culture and identification.
Detailed Protocol Steps [108] [109] [80]:
- Sample Collection and Processing: Samples (e.g., bronchoalveolar lavage (BAL), sputum, wound debridement) are collected aseptically. Samples may be homogenized and serially diluted in a buffered solution [109] [80].
- Inoculation and Incubation: Processed samples are inoculated onto appropriate agar plates (e.g., blood agar, chocolate agar, MacConkey agar) to support the growth of different types of microorganisms. The plates are then incubated under controlled conditions (e.g., 35±2°C for 24-48 hours) to allow for the development of visible colonies [108].
- Colony Identification: Visible colonies are counted and subcultured to obtain pure isolates. Identification is performed using phenotypic methods such as Gram staining, analysis of colony morphology, and biochemical tests (e.g., catalase, oxidase, IMViC tests) [108] [80]. Modern labs may use MALDI-TOF mass spectrometry for identification from pure colonies [108].
- Antimicrobial Susceptibility Testing (AST): For bacterial isolates, AST is performed using methods like disk diffusion, broth microdilution, or automated systems (e.g., Vitek 2) to determine the minimum inhibitory concentration (MIC) of antimicrobial agents [108].
Rapid Molecular Method (PCR-Based Panel) Protocol
Rapid molecular methods, such as multiplex PCR panels, detect microbial nucleic acids. The following diagram illustrates a typical workflow for a PCR-based panel like the BioFire Pneumonia Panel.
Detailed Protocol Steps [108]:
- Sample Preparation: The patient sample (BAL, tracheal aspirate, or sputum) is directly inoculated into a provided buffer solution within a loading pouch [108].
- Automated Nucleic Acid Testing: The loaded pouch is inserted into the instrument (e.g., BioFire Torch). The system automates all subsequent steps:
- Nucleic Acid Extraction: The target nucleic acids (DNA and RNA) are released and purified from the sample.
- Multiplex PCR Amplification: The purified nucleic acids are subjected to a multiplex PCR reaction, which simultaneously amplifies targets for a comprehensive panel of bacteria, viruses, and antimicrobial resistance genes.
- Detection and Analysis: Amplification products are detected in real time using high-resolution melt curve analysis. The instrument calculates approximate concentrations for quantitative bacterial targets and provides a final report in about an hour [108].
Analysis of Concordance and Discrepancies
The comparison between traditional and rapid methods reveals patterns of strong concordance but also critical, clinically relevant discrepancies.
Areas of High Concordance
- Organism Identification: When an organism is detected by both methods, identification is highly concordant. One study on pneumonia pathogens reported 97.4% concordance between the BioFire PN Panel and traditional culture for organism identification across thousands of data points [108].
- Antimicrobial Resistance (AMR) Marker Detection: Detection of genetic resistance markers shows excellent agreement with phenotypic resistance. The same study found 99% concordance for the detection of resistance genes (e.g., mecA, blaKPC) when compared to various culture-based AST methods [108].
Nature and Causes of Discrepancies
Despite high concordance in some areas, significant discrepancies exist, primarily due to the fundamental differences in what each method detects.
- Increased Sensitivity of Molecular Methods: Molecular methods consistently detect a greater number and diversity of organisms. A study on chronic wounds found that culture identified only 17 different bacterial taxa, whereas 16S rDNA sequencing identified 338 different taxa [109]. This is because PCR can detect:
- Fastidious or Slow-Growing Organisms that may not thrive on standard culture media [108].
- Viable But Non-Culturable (VBNC) Organisms that are metabolically active but cannot form colonies on a plate [80].
- Low Bioburden Samples where the number of organisms is below the detection limit of culture [1].
- Correlation with Microbial Load: The semiquantitative data from rapid PCR panels can help interpret discrepancies. One study demonstrated a direct relationship between the PCR result (in copies/mL) and the probability of cultural growth. Only 4% of organisms detected at 10^4 copies/mL grew in culture, compared to 53% of those detected at 10^7 copies/mL [108]. This suggests that low-level signals from molecular methods may represent colonization or non-viable organisms rather than active infection.
- Detection of Non-Viable Organisms: A key limitation of nucleic acid-based methods is their inability to distinguish between viable and non-viable cells, as they detect DNA from both live and dead microorganisms. In contrast, culture only detects viable, culturable organisms [80]. This can lead to "over-detection" by molecular methods in samples containing non-viable organisms from prior infections or antimicrobial treatment.
The Scientist's Toolkit: Essential Research Reagents and Materials
Selecting the appropriate reagents and materials is fundamental for successfully implementing either traditional or rapid microbiological methods. The following table details key solutions used in the featured experiments.
Table 2: Essential Research Reagent Solutions for Microbial Detection
| Reagent/Material | Function | Application Context |
|---|---|---|
| Selective Culture Media (e.g., Blood Agar, MacConkey Agar) | Supports growth of specific microorganism types while inhibiting others. | Traditional Culture [108] |
| BioFire Pneumonia Panel | Integrated pouch containing reagents for nucleic acid extraction, amplification, and detection of pneumonia pathogens. | Rapid Molecular PCR [108] |
| Lysis Buffer (e.g., RLT Buffer with β-mercaptoethanol) | Disrupts cells to release nucleic acids for downstream molecular analysis. | DNA Extraction for Sequencing [109] |
| DNA Spin Columns (e.g., QIAamp DNA Mini Kit) | Purifies nucleic acids from sample lysates by binding DNA and removing contaminants. | DNA Extraction for Sequencing [109] |
| Primers for 16S rDNA (e.g., 28F/519R) | Amplifies a conserved region of the bacterial 16S rRNA gene for identification and sequencing. | 16S rDNA Sequencing [109] |
| Multiplex PCR Master Mix | Contains enzymes, dNTPs, and buffers for simultaneous amplification of multiple DNA targets. | Rapid Molecular PCR [108] |
The data presented in this guide objectively demonstrates that both traditional culture and rapid microbiological methods have distinct roles in contamination detection and clinical diagnostics. Traditional methods provide a proven, low-tech means of obtaining viable isolates for further phenotypic testing, such as AST. However, they are limited by longer turnaround times and lower sensitivity, particularly for fastidious organisms or complex polymicrobial samples [108] [109] [80].
Rapid molecular methods, particularly PCR-based panels, offer significant advantages in speed, sensitivity, and breadth of detection. They enable more informed and timely clinical decisions, potentially improving patient outcomes and facilitating antibiotic stewardship [108] [80]. The high clinical concordance for identified pathogens and resistance markers supports their reliability.
The choice between methods should not be viewed as a binary decision but rather as a strategic one. For clinical diagnostics, a synergistic approach that leverages the speed and sensitivity of molecular methods for initial diagnosis, complemented by the viability data and isolate generation from culture for confirmed infections, is often the most effective strategy. Furthermore, the validation of rapid methods against international standards, as overseen by bodies like MicroVal and PDA, ensures their reliability and promotes their wider adoption in regulated industries [111] [110] [72]. Researchers and clinicians must interpret results within the specific clinical or manufacturing context, understanding the inherent strengths and limitations of each technological paradigm.
Conclusion
The choice between traditional and rapid microbiological methods is not a simple binary but a strategic decision informed by application, regulatory requirements, and operational goals. While traditional methods remain a validated, cost-effective standard, rapid methods offer transformative advantages in speed, sensitivity, and automation, crucial for accelerating drug development and enhancing contamination control. Future directions point toward greater integration of AI and machine learning for predictive analysis, the development of portable point-of-care devices, and the ongoing need for standardized protocols to ensure reliability. For biomedical research and clinical practice, the strategic adoption and validation of RMMs are pivotal for advancing antimicrobial stewardship, managing complex infections, and ultimately delivering safer pharmaceuticals to market faster.
References
- 1. Comparing Traditional vs. Rapid Microbial Testing [https://puroxi.com/traditional-vs-rapid-microbial-testing/]
- 2. Principles of microscopy, culture and serology-based ... [https://www.sciencedirect.com/science/article/abs/pii/S1357303921002012]
- 3. Conventional methods and future trends in antimicrobial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9958398/]
- 4. Traditional and Molecular Techniques for the Study of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3369584/]
- 5. Comparison of Rapid and Routine Methods of Identification ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7324860/]
- 6. A comparison of surface sampling methods for detecting ... [https://www.sciencedirect.com/science/article/pii/S0740002001904642]
- 7. Editorial on Rapid vs Traditional Microbiological Methods ... [https://wickhammicro.co.uk/knowledge-and-education/editorial-on-rapid-vs-traditional-microbiological-methods-published-in-ipi]
- 8. An Overview of Rapid Microbial-Detection Methods [https://www.pharmtech.com/view/overview-rapid-microbial-detection-methods]
- 9. Article 1 Rapid microbiological methods 2011 [https://www.europeanpharmaceuticalreview.com/article/5622/article-1-rapid-microbiological-methods-2011/]
- 10. The RMM Product Matrix - Rapid Microbiological Methods [https://rapidmicromethods.com/files/matrix.php]
- 11. Molecular Methods for Pathogenic Bacteria Detection and ... [https://www.mdpi.com/2073-4441/13/24/3551]
- 12. A review on detection methods used for foodborne ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5320838/]
- 13. "Emerging technologies for detecting foodborne pathogens ... [https://www.sciencedirect.com/science/article/abs/pii/S0740002025000437]
- 14. A Comprehensive Approach to Testing with Microorganisms [https://www.americanpharmaceuticalreview.com/Featured-Articles/615654-Evaluating-Rapid-Microbial-Methods-A-Comprehensive-Approach-to-Testing-with-Microorganisms/]
- 15. Performance Comparison of Rapid Petrifilm™ Test ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7259363/]
- 16. Advancing microbial risk assessment: perspectives from ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12304167/]
- 17. The Barriers to RMM Adoption [https://www.rapidmicrobio.com/learning-center/rapid-blog/the-barriers-to-rmm-adoption]
- 18. AI driving automated microbiological testing market growth ... [https://www.europeanpharmaceuticalreview.com/news/264463/ai-driving-automated-microbiological-testing-market-growth-to-2033/]
- 19. AI trends 2025: Adoption barriers and updated predictions [https://www.deloitte.com/us/en/what-we-do/capabilities/applied-artificial-intelligence/blogs/pulse-check-series-latest-ai-developments/ai-adoption-challenges-ai-trends.html]
- 20. Mapping research trends in milk microbiology: Rapid ... [https://www.sciencedirect.com/science/article/pii/S0023643825011624]
- 21. Remote Monitoring & Management (RMM) Tools 2025- ... [https://www.archivemarketresearch.com/reports/remote-monitoring-management-rmm-tools-51801]
- 22. An Overview on Novel Microbial Determination Methods in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5071793/]
- 23. Real-Time PCR (qPCR) and Whole-Transcriptome NGS [https://www.thermofisher.com/us/en/home/life-science/pcr/real-time-pcr/ngs-comparison.html]
- 24. Sensitive, real-time PCR detects low-levels of ... [https://www.sciencedirect.com/science/article/abs/pii/S0890850805000770]
- 25. Application of next-generation sequencing for detecting ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12549248/]
- 26. Common Contaminants in Next-Generation Sequencing That ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0097876]
- 27. Comparison of Next-Generation Sequencing, Real-Time ... [https://www.mdpi.com/2076-2607/13/10/2344]
- 28. Real-time PCR and targeted next-generation sequencing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6656700/]
- 29. Combating viral contamination and disease outbreaks at ... [https://nanoporetech.com/resource-centre/combating-viral-contamination-and-disease-outbreaks-at-source-with-rapid-low-cost-nanopore-sequencing]
- 30. Performance Comparison of Droplet Digital PCR and Next ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12062871/]
- 31. Assessment and Comparison of Viability Assays for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10872314/]
- 32. Measuring Cytotoxicity by Bioluminescence Imaging ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0089357]
- 33. Novel method detects microbial contamination in cell cultures [https://news.mit.edu/2025/novel-method-detects-microbial-contamination-smart-0425]
- 34. A Systematic Comparison Identifies an ATP-Based Viability ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4871979/]
- 35. Using the ATP luminescence-based method to determine ... [https://www.nature.com/articles/s41598-025-03682-5]
- 36. Comparison of Three Luciferase-Based Detection Kits for ... [https://www.bioagilytix.com/resources/posters/comparison-of-three-luciferase-based-detection-kits-for-cell-viability-assessment-in-a-cell-based-potency-assay/]
- 37. Comparison of Different Methods to Measure Cell Viability [https://www.creative-bioarray.com/support/comparison-of-different-methods-to-measure-cell-viability.htm]
- 38. Comparison of the ATP-bioluminescence assay and ... [https://tmuj.iautmu.ac.ir/browse.php?a_id=2070&sid=1&slc_lang=en]
- 39. The Four Key Benefits of Rapid Micro Methods - bioMerieux [https://www.biomerieux.com/corp/en/blog/pharmaceutical-qc/the-four-key-benefits-of-rmm.html]
- 40. Mass Spectrometry-Based Biosensing and Biopsy Technology [https://www.mdpi.com/2227-9040/11/8/419]
- 41. Surface mass spectrometry — application to biosensor characterization - ScienceDirect [https://www.sciencedirect.com/science/article/abs/pii/095656639599226B]
- 42. Biosensors, Volume 15, Issue 9 (September 2025) [https://www.mdpi.com/2079-6374/15/9]
- 43. Contaminant Identification in Pharmaceutical Products [https://www.mccrone.com/mm/contaminant-identification-pharmaceutical-products/]
- 44. Challenges of growth-based microbiological methods in ... [https://link.springer.com/article/10.1007/s44395-025-00020-6]
- 45. Microbiology/RMM In-Depth Focus 2025 [https://www.europeanpharmaceuticalreview.com/article/265182/microbiology-rmm-in-depth-focus-2025/]
- 46. Automated plate reading start-up lands AstraZeneca and ... [https://cleanroomtechnology.com/automated-plate-reading-start-up-lands-astrazeneca-and]
- 47. 5 Characteristics Of Forward-Thinking Microbiology Labs In ... [https://www.pharmaceuticalonline.com/doc/characteristics-of-forward-thinking-microbiology-labs-in-2025-0001]
- 48. Translating microbial kinetics into quantitative responses ... [https://www.nature.com/articles/s41467-025-61592-6]
- 49. Using AI to screen clinical microbiology plates [https://www.rapidmicrobiology.com/supplier/clever-culture-systems]
- 50. Performance Survey and Comparison Between Rapid ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5816116/]
- 51. Bioburden Testing & Rapid Microbiology Methods [https://www.watertechnologies.com/applications/bioburden]
- 52. Implementing Rapid Sterility Test Systems: A Guide from ... [https://www.criver.com/eureka/implementing-rapid-sterility-test-systems-guide-experts]
- 53. Comparison of Statistical Process Control Models for ... [https://www.mdpi.com/2227-9717/13/9/2917]
- 54. Detection limitations of bacteria in tissue samples - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12178716/]
- 55. Rapid Microbial Detection Relies on Traditional Media Power [https://www.rapidmicrobiology.com/news/rapid-microbial-detection-relies-on-the-power-of-traditional-media]
- 56. Biofilms Exposed: Innovative Imaging and Therapeutic ... [https://www.mdpi.com/2079-6382/14/9/865]
- 57. New Methodologies as Opportunities in the Study of Bacterial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12114119/]
- 58. A novel dual-staining method for cost-effective visualization ... [https://www.nature.com/articles/s41598-024-80644-3]
- 59. Effect of biofilm physical characteristics on their ... [https://www.nature.com/articles/s41522-024-00544-2]
- 60. Label-free microscopy enables high-throughput identification ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12424827/]
- 61. Emerging Technologies and Integrated Strategies for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12299165/]
- 62. Comprehensive Microbial Control Strategy [https://www.bioprocessintl.com/qa-qc/beyond-traditional-microbiology-testing-developing-a-comprehensive-strategy-for-microbial-control-and-detection]
- 63. Viable but nonculturable [https://en.wikipedia.org/wiki/Viable_but_nonculturable]
- 64. Viable but Nonculturable - an overview [https://www.sciencedirect.com/topics/immunology-and-microbiology/viable-but-nonculturable]
- 65. Current Perspectives on Viable but Non-Culturable ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10048424/]
- 66. The Significance and Detection of VBNC Microorganisms [https://www.americanpharmaceuticalreview.com/Featured-Articles/113051-The-Significance-and-Detection-of-VBNC-Microorganisms/]
- 67. Latest Developments of Research on the Viable Non ... [https://www.mdpi.com/2076-3417/15/3/1454]
- 68. Comparison of Standard and Impedance Methods for the ... [https://brieflands.com/journals/jjnpp/articles/64341]
- 69. Modern Microbial Methods Supporting a Contamination ... [https://www.pda.org/pda-letter-portal/home/full-article/modern-microbial-methods-support-of-a-contamination-control-strategy]
- 70. Absolute quantification of VBNC state formation and ... [https://pubmed.ncbi.nlm.nih.gov/41122686/]
- 71. A comparison of traditional and recently developed ... [https://pubmed.ncbi.nlm.nih.gov/12590780/]
- 72. Rapid Microbiological Methods for Pharmaceutical ... [https://www.rapidmicrobiology.com/test-method/rapid-microbiological-methods-for-pharmaceutical-laboratories]
- 73. Innovative analytical methods for monitoring ... [https://www.sciencedirect.com/science/article/abs/pii/S0026265X19303613]
- 74. Improving Turnaround Times for Routine Antimicrobial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11591232/]
- 75. Microbiological methodologies: Comparative evaluation of ... [https://www.sciencedirect.com/science/article/pii/S0717345825000053]
- 76. Breaking the Rapid Microbiological Method Financial Barrier [https://www.biopharminternational.com/view/breaking-rapid-microbiological-method-financial-barrier-case-study-return-investment-and-economic-ju]
- 77. Return on Investment (ROI) - Rapid Microbiological Methods [http://rapidmicromethods.com/files/roi.php]
- 78. Pharmaceutical Rapid Microbiology Testing Market [https://www.novaoneadvisor.com/report/pharmaceutical-rapid-microbiology-testing-market]
- 79. 5 Limitations of Traditional Microbial Testing [https://www.rapidmicrobio.com/learning-center/rapid-blog/5-limitations-of-traditional-microbial-testing-]
- 80. A Comprehensive Review of Innovative Paradigms in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11216122/]
- 81. Review Articles Validation of Rapid Microbiological Methods [https://www.sciencedirect.com/science/article/pii/S2472630322014698]
- 82. NIST Rapid Microbial Testing Methods Consortium [https://www.nist.gov/programs-projects/nist-rapid-microbial-testing-methods-consortium]
- 83. Rapid Microbiological Methods in Quality Control of Sterile ... [https://www.mabion.eu/science-hub/articles/rapid-microbiological-methods-in-quality-control-of-sterile-drugs/]
- 84. 微生物质量控制-制药qc实验室-检测方法 [https://www.sartorius.com.cn/applications/quality-control-testing/microbiological-quality-control]
- 85. Performance Equivalence and Validation of a Rapid ... [https://pubmed.ncbi.nlm.nih.gov/37085183/]
- 86. Mastering Quality: New Method Validation, QC Standards, PT [https://www.rapidmicrobiology.com/news/mastering-quality-new-method-validation-qc-standards-and-pt-a-rapidmicrobiology-special-focus]
- 87. Rapid microbiological methods and the regulatory ... [https://www.europeanpharmaceuticalreview.com/article/81089/rapid-microbiological-methods/]
- 88. Empowering your GMP workflow with Quality Control ... [https://www.rapidmicrobio.com/learning-center/rapid-blog/empowering-your-gmp-workflow-with-quality-control-automation]
- 89. . Ph ., Eur and other Pharmacopoeias - Live Online Training - ECA... USP [https://www.gmp-compliance.org/training/gmp-course-conference/ep-usp-and-other-pharmacopoeias]
- 90. Validate Rapid Microbiological Methods for ... [https://www.pharmanow.live/regulatory-landscape/validate-rapid-microbiological-methods]
- 91. analytics-shop.com/gb/hplc-analysis- usp - ph - eur [https://www.analytics-shop.com/gb/hplc-analysis-usp-ph-eur]
- 92. Harmonization Status for General Methods [https://www.usp.org/harmonized-standards/pdg/general-methods]
- 93. What are the differences and key steps in Analytical ... [https://resources.eirgenix.com/blog/method_dev_qual_validation]
- 94. Application of 'robust' methods to the analysis ... [https://www.sciencedirect.com/science/article/abs/pii/S0167701206000364]
- 95. A step by step guide to quantitative comparisons in ... [https://finbiosoft.com/a-step-by-step-guide-to-quantitative-comparisons-in-validation-manager/]
- 96. A Guide to USP <1223> and Validation of Rapid Methods [https://www.americanpharmaceuticalreview.com/Featured-Articles/613587-Validating-Alternative-Microbiological-Methods-A-Guide-to-USP-1223-and-Validation-of-Rapid-Methods/]
- 97. The Essential Guide to Microbiology Testing: Methods, ... [https://contractlaboratory.com/microbiology-testing-services/]
- 98. A Review of Modern Methods for the Detection ... [https://www.mdpi.com/2076-2607/11/5/1111]
- 99. validation and Method verification method [https://committee.iso.org/sites/tc34sc9/home/essential-information/content-left-area/validation-of-methods/method-validation-and-method-ver.html]
- 100. An approach for interlaboratory of conventional and... comparison [https://pubmed.ncbi.nlm.nih.gov/23562705/]
- 101. – Interlaboratory degree of comparisons demonstrating equivalence [https://link.springer.com/article/10.1007/s00769-003-0580-5]
- 102. sciencedirect.com/topics/earth-and-planetary-sciences/ interlaboratory ... [https://www.sciencedirect.com/topics/earth-and-planetary-sciences/interlaboratory-comparison]
- 103. ISO 16140-3:2021 Method Verification Roundtable [https://www.neogen.com/en/neocenter/resources/iso-16140-32021-method-verification-roundtable-perspectives-from-the-industry/]
- 104. What You Need to Know About the New ISO 16140-3 ... [https://www.sigmaaldrich.com/US/en/collections/webinars/iso-16140-3-microbiological-method-verification?srsltid=AfmBOoo2U9FoNWIb5r1IpZVC4c8EYNSXej4R19bDuvxb1Xou6jMq3j6w]
- 105. Contamination Detection in Pharma Products Market 2025 ... [https://www.towardshealthcare.com/insights/contamination-detection-in-pharma-products-market-sizing]
- 106. Comparison of Microbiological and High-Performance ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3862973/]
- 107. Comparative performance of contact plate metod and swab ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11129454/]
- 108. Clinical utilization and culture concordance of categorical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12026127/]
- 109. Comparison of Culture and Molecular Identification ... [https://www.mdpi.com/1422-0067/13/3/2535]
- 110. PDA Journal of Pharmaceutical Science and Technology [https://journal.pda.org/content/early/2023/04/21/pdajpst.2021.012632]
- 111. AOAC & MicroVal Method Validation Training [https://microval.org/2025/09/02/aoac-microval-method-validation-training-microbial-method-validation-detailed-insights/]