Proximity Labeling Enzymes Compared: A Guide to Intracellular Tagging for Biomedical Research
Proximity labeling (PL) has revolutionized the study of protein interactions and subcellular proteomes in living systems.
Proximity Labeling Enzymes Compared: A Guide to Intracellular Tagging for Biomedical Research
Abstract
Proximity labeling (PL) has revolutionized the study of protein interactions and subcellular proteomes in living systems. This article provides a comprehensive comparison of PL enzymes, from foundational tools like BioID and APEX to recent innovations such as TurboID and light-activated systems. We detail their mechanisms, key applications across model organisms, and method-specific advantages for capturing dynamic biological processes. A dedicated troubleshooting section addresses common challenges like background labeling and experimental variation. By synthesizing validation strategies and comparative performance data, this guide empowers researchers and drug development professionals to select and optimize the right PL tool for their specific intracellular tagging needs, from basic research to target discovery.
The Engine of Discovery: Understanding Proximity Labeling Enzyme Families and Mechanisms
Enzymatic proximity labeling is a powerful technique in molecular biology that enables researchers to map the spatial organization and interaction networks of proteins and nucleic acids within their native cellular environment. By fusing an enzyme to a protein of interest, scientists can tag nearby biomolecules with a biotin label, allowing for their subsequent isolation and identification. This guide compares the performance, applications, and experimental protocols for the primary enzymatic systems used in intracellular tagging.
Fundamental Mechanisms of Proximity Labeling
The core principle of proximity labeling involves genetically fusing a "bait" protein of interest to a specialized enzyme. When a small, cell-permeable substrate is added, the enzyme generates highly reactive, short-lived molecules that covalently tag nearby "prey" proteins within a limited radius. These biotin-tagged proteins can then be purified under stringent denaturing conditions using streptavidin beads and identified through mass spectrometry, providing a snapshot of the local proteomic environment [1] [2].
The two primary enzyme classes used are biotin ligases and peroxidases, which operate through distinct catalytic mechanisms:
Biotin Ligases (e.g., BioID, TurboID) use ATP to activate biotin into a reactive biotinoyl-5'-AMP (bioAMP) intermediate. In engineered promiscuous variants, this intermediate is released and covalently attaches to lysine residues on proximate proteins [1] [3].
Peroxidases (e.g., APEX/APEX2) use hydrogen peroxide (H₂O₂) to oxidize biotin-phenol into a highly reactive phenoxyl radical that rapidly tags tyrosine residues on nearby proteins. This reaction occurs within a very short time frame (as little as 1 minute) [4] [2].
Comparative Analysis of Proximity Labeling Enzymes
The following table summarizes the key characteristics of the most commonly used proximity labeling enzymes, highlighting critical differences in speed, size, and operational requirements.
Table 1: Key Characteristics of Proximity Labeling Enzymes
| Enzyme | Size (kDa) | Labeling Time | Key Substrate | Primary Residue Labeled | Key Advantages | Major Limitations |
|---|---|---|---|---|---|---|
| BioID [1] [3] | 35 | 18-24 hours | Biotin | Lysine | Low background, non-toxic | Very slow kinetics |
| BioID2 [3] [2] | ~25 | >16 hours | Biotin | Lysine | Smaller size, improved localization | Still requires long labeling times |
| TurboID [3] [2] | 35 | 10 minutes | Biotin | Lysine | Extremely fast, high sensitivity | Can be toxic at high expression levels, higher background |
| miniTurbo [3] | 28 | 10 minutes | Biotin | Lysine | Fast, smaller size, lower pre-labeling background | Slightly less active than TurboID |
| APEX/APEX2 [4] [2] | 28 | 1 minute | Biotin-phenol + H₂O₂ | Tyrosine | Fastest labeling, works in multiple organelles | H₂O₂ is toxic, can cause oxidative stress |
Performance and Experimental Data
Direct comparisons of these enzymes reveal significant performance differences. In HEK 293T cells, TurboID and miniTurbo biotinylated endogenous proteins much more rapidly than BioID, producing a 3-6 fold difference in signal at early time points and a 15-23 fold difference at later time points [3]. TurboID generated as much biotinylated product in 10 minutes as BioID achieved in 18 hours [3].
The following performance data table synthesizes quantitative findings from comparative studies:
Table 2: Experimental Performance Metrics of Proximity Labeling Enzymes
| Enzyme | Relative Activity vs. BioID | Minimum Detectable Labeling Time | Toxicity Concerns | Optimal Applications |
|---|---|---|---|---|
| TurboID | ~15-23x higher (18-hour comparison) [3] | <10 minutes [3] | Yes (can affect cell viability) [2] | Rapid processes, low-expression baits, in vivo models [3] [2] |
| miniTurbo | ~10-15x higher (18-hour comparison) [3] | <10 minutes [3] | Minimal [3] | Rapid processes requiring precise temporal control [3] |
| APEX2 | N/A (different mechanism) | ~1 minute [2] | Yes (H₂O₂-induced oxidative stress) [2] | Ultrarapid processes, electron microscopy, RNA labeling [4] [2] [5] |
| BioID | Baseline | ~18 hours [1] [3] | Minimal [2] | Stable complexes, high-expression baits, non-toxic requirement [1] |
Advanced Applications and Specialized Systems
Split-System and Condition-Activated Labeling
Advanced engineering has produced more precise systems that activate only under specific conditions. The split-TurboID system separates the enzyme into two inactive fragments that only reconstitute and become active when the proteins they're fused to interact closely. This significantly reduces background labeling and enables the validation of specific protein-protein interactions [2] [6].
A notable innovation is Ca²⁺-activated split-TurboID (CaST), which acts as a coincidence detector for both exogenous biotin and elevated intracellular calcium. This system tethers the split-TurboID fragments to calmodulin (CaM) and an M13 peptide. At high Ca²⁺ concentrations, CaM and M13 interact, reconstituting TurboID activity and biotinylating nearby proteins. This allows researchers to "record" calcium signaling events in neurons and other cell types with a temporal resolution of approximately 10 minutes [6].
RNA-Protein Interaction Mapping
Proximity labeling has been adapted to study RNA-protein interactions through methods like HyPro (Hybridization-based Proximity Labeling). In this approach, a fixed and permeabilized enzyme (typically an APEX2 derivative) is targeted to specific RNA molecules using digoxigenin-labeled antisense oligonucleotides. The enzyme then biotinylates proteins associated with the RNA, enabling proteomic profiling of specific ribonucleoprotein complexes. Recent enhancements (HyPro2) have improved labeling efficiency for low-abundance RNA targets, including single RNA molecules [4].
Essential Research Reagent Solutions
Successful proximity labeling experiments require carefully selected reagents and optimization. The following table outlines key solutions and their applications:
Table 3: Essential Research Reagent Solutions for Proximity Labeling
| Reagent / Solution | Function | Application Notes |
|---|---|---|
| Biotin | Substrate for biotin ligases (BioID, TurboID) | Use 50-500 μM for TurboID/miniTurbo; cell-permeable and non-toxic [3] |
| Biotin-Phenol | Substrate for peroxidases (APEX/APEX2) | Converted to phenoxyl radical by H₂O₂; tags tyrosine residues [4] [2] |
| Hydrogen Peroxide (H₂O₂) | Oxidizing agent for APEX/APEX2 | Required for peroxidase activity; can cause cellular stress - optimize concentration [2] |
| Streptavidin Beads | Affinity purification of biotinylated proteins | High affinity for biotin; enables stringent washing to reduce contaminants [1] [3] |
| Tyramide Signal Amplification (TSA) | Signal enhancement system | Can boost biotin signal in low-activity systems [3] |
| Trehalose | Viscosity-modifying compound | Reduces diffusion of reactive species, improving spatial resolution (50% in labeling buffer) [4] |
Experimental Protocol: TurboID Labeling in Mammalian Cells
The following protocol outlines a standard TurboID experiment for mapping protein interactions in living mammalian cells, based on established methodologies [3]:
Construct Design: Fuse TurboID to your protein of interest using standard molecular cloning techniques. Include a flexible linker (e.g., GGGS repeats) between the protein and TurboID to minimize steric interference.
Cell Transfection and Expression: Transfect HEK 293T or other relevant cell lines with your TurboID fusion construct. Allow 24-48 hours for protein expression. Validate proper localization using microscopy if possible.
Biotin Labeling: Add biotin to the culture medium at a final concentration of 50-500 μM. For TurboID, incubate for 10 minutes to several hours depending on the desired labeling depth. Critical Note: Include appropriate controls (e.g., untransfected cells, non-fused TurboID expressed in the same compartment).
Termination and Cell Lysis: Remove biotin-containing medium and wash cells thoroughly with cold phosphate-buffered saline (PBS) to stop the reaction. Lyse cells using RIPA buffer or similar denaturing lysis buffer containing protease inhibitors.
Streptavidin Affinity Purification: Incubate clarified cell lysates with streptavidin-coated beads for 1-2 hours at room temperature. Wash beads stringently with lysis buffer, high-salt buffer, and carbonate buffer to reduce non-specific binding.
On-Bead Digestion and Proteomic Analysis: Digest biotinylated proteins on beads using trypsin. Desalt peptides and analyze by liquid chromatography with tandem mass spectrometry (LC-MS/MS).
Data Analysis: Process MS data using standard proteomic software. Compare experimental samples against controls to identify significantly enriched proteins that represent genuine proximate interactors.
Enzymatic proximity labeling has revolutionized the study of spatial proteomics in living cells. The choice between biotin ligases (BioID, TurboID, miniTurbo) and peroxidases (APEX/APEX2) depends on experimental priorities: TurboID offers unprecedented speed for dynamic processes, while APEX2 provides the highest temporal resolution for rapid events. BioID remains valuable for its minimal toxicity despite slower kinetics. Recent innovations like split-systems and condition-activated enzymes are expanding applications to include mapping transient signaling events and validating specific protein interactions. As these technologies continue to evolve, they will provide increasingly sophisticated tools for deciphering the complex molecular architecture of living systems.
In the field of intracellular proximity labeling, temporal resolution—the ability to capture molecular interactions at specific moments in time—is a critical parameter for studying dynamic cellular processes. Among peroxidase-based tagging tools, APEX (Ascorbate Peroxidase) and its engineered descendant APEX2 stand out for their exceptional speed, enabling researchers to obtain snapshots of proteomic landscapes within minute-scale time windows. This guide provides an objective comparison of their performance, focusing on the key metrics of labeling speed, efficiency, and temporal control that are vital for research and drug development.
Head-to-Head: APEX vs. APEX2 at a Glance
The following table summarizes the core characteristics of APEX and APEX2, highlighting the key improvements that define their performance in experimental settings.
| Feature | APEX | APEX2 |
|---|---|---|
| Origin | Engineered from plant ascorbate peroxidase (APX) [7] | Directed evolution of APEX for enhanced activity and stability [8] |
| Labeling Time | ~1 minute [8] | ~1 minute [9] |
| Primary Advantage | Rapid labeling; functions in the reducing cytosolic environment [7] | Higher activity, improved signal-to-noise ratio, and better expression at low levels [8] |
| Key Structural Traits | Lacks disulfide bonds and calcium dependence; ~28 kDa [7] [10] | Same robust structural traits as APEX [10] |
| Cytotoxicity Concerns | Requires mM H(2)O(2), which can be toxic [11] | Same H(2)O(2) requirement; toxicity is a shared challenge [11] |
Quantitative Performance and Experimental Data
Catalytic Efficiency and Stability
A biophysical comparison between horseradish peroxidase (HRP) and APEX underscores the stability of the APEX system. Molecular dynamics simulations revealed that the split form of APEX2 (sAPEX2) exhibited the smallest structural variations, indicating superior stability compared to other peroxidases. This intrinsic stability is a key factor supporting its consistent performance in diverse cellular compartments [10].
Direct Comparison of Labeling Specificity
A 2021 study profiling striatal neuron proteomes demonstrated APEX2's effectiveness ex vivo. The workflow provided sufficient depth to uncover activity-dependent changes in the proteome following chemogenetic activation, showcasing its capability for precise, time-resolved snapshots [9]. The high specificity of APEX2-mediated labeling in this complex tissue context underscores its utility in challenging experimental systems.
Detailed Experimental Protocols
To ensure reproducibility, below are detailed methodologies for key applications of APEX and APEX2 as cited in the literature.
This protocol is designed for cell-type-specific subcellular proteomics in mouse brain tissue.
- Viral Transduction: Neonatal mice are virally transduced with Cre-dependent AAVs encoding the APEX2 construct fused to a subcellular targeting sequence (e.g., H2B for nucleus, NES for cytosol, LCK for membrane).
- Tissue Preparation: After 5-6 weeks, prepare 250 µm acute brain slices in carbogenated artificial cerebrospinal fluid (ACSF).
- Biotinylation:
- Incubation: Incubate slices in ACSF supplemented with 500 µM Biotin-Phenol (BP) for 1 hour to allow substrate penetration.
- Activation: Transfer slices to ACSF containing 0.03% H(2)O(2) for 1 minute to induce labeling.
- Quenching: Immediately immerse slices in a quenching solution containing Trolox, sodium ascorbate, and sodium azide to stop the reaction.
- Tissue Dissection and Processing: Dissect the region of interest based on a fluorescent reporter, then lyse the tissue for streptavidin-based enrichment and mass spectrometry analysis.
This 2025 protocol addresses H(2)O(2) toxicity and background labeling by using an enzymatic cascade to generate H(2)O(2) in situ.
- Cell Line Generation: Create a stable cell line expressing both a target protein (e.g., ciliary protein NPHP3) fused to APEX2 and a second target protein fused to D-amino acid oxidase (DAAO).
- Labeling Induction: Instead of adding H(2)O(2) directly, add the DAAO substrate D-Alanine (1-5 mM) to the culture medium for 30 minutes. DAAO locally produces H(2)O(2), which is immediately used by the nearby APEX2.
- Biotinylation: Simultaneously with D-Alanine addition, include Biotin Tyramide (BT) to enable proximity labeling.
- Analysis: Proceed with cell lysis, streptavidin pull-down, and mass spectrometry. This method minimizes non-specific background and expands APEX2 use to cell types previously incompatible with conventional labeling [11].
The Scientist's Toolkit: Essential Research Reagents
The table below lists key reagents required for conducting APEX/APEX2 proximity labeling experiments.
| Reagent / Tool | Function / Description | Key Consideration |
|---|---|---|
| APEX/APEX2 Constructs | Genetically encoded enzyme, fused to a protein or localization signal of interest. | APEX2 is preferred for its higher activity and stability [8]. |
| Biotin-Phenol (BP) | Enzyme substrate. Oxidized to a phenoxyl radical that tags nearby proteins. | The radical has a short diffusion radius (~20 nm), ensuring high spatial resolution [8]. |
| Hydrogen Peroxide (H₂O₂) | Electron acceptor required to activate the peroxidase. | High concentrations (mM) can be cytotoxic; timing is critical [11]. |
| DAAO & D-Amino Acids | Alternative, less toxic system for H₂O₂ generation in the iAPEX workflow. | DAAO from Rhodotorula gracilis is specific for D-amino acids, reducing off-target effects [11]. |
| Quenching Solution | A solution (e.g., containing Trolox, sodium ascorbate) to stop the labeling reaction. | Essential for achieving precise temporal control [9]. |
| Streptavidin Beads | High-affinity resin for purifying biotinylated proteins prior to mass spectrometry. | Allows for stringent washing to reduce background noise. |
Operational Workflow and Novel Applications
The following diagram illustrates the core operational workflow of APEX2 proximity labeling, from genetic targeting to proteomic analysis.
A recent innovative application, the iAPEX (in situ APEX activation) system, tackles the limitation of H(2)O(2) toxicity. This system co-localizes APEX2 with the enzyme D-amino acid oxidase (DAAO). Upon adding a D-amino acid (e.g., D-Alanine), DAAO produces H(2)O(2) locally, which is immediately used by APEX2 for labeling. This enzymatic cascade minimizes background and expands the technology's use to more sensitive biological systems [11].
APEX and APEX2 provide unparalleled temporal resolution in the proximity labeling toolkit. The primary distinction lies in APEX2's enhanced catalytic activity and stability, which translates directly to higher labeling efficiency and a better signal-to-noise ratio in experimental data [8]. This makes APEX2 the preferred choice for most contemporary applications, particularly when working with low-abundance targets or complex tissues like the brain [9].
A shared consideration for both enzymes is the potential cytotoxicity of exogenous H(2)O(2). The emerging iAPEX system, which uses DAAO to generate H(2)O(2) in situ, presents a powerful solution. It not only reduces toxicity but also significantly improves specificity by minimizing background labeling from endogenous peroxidases, thereby expanding the potential for in vivo applications [11].
In summary, for researchers investigating rapid, dynamic intracellular processes—from neuronal activity-dependent changes to receptor internalization pathways—APEX2 currently offers the optimal combination of speed, precision, and robustness. The ongoing development of refined systems like iAPEX promises to further broaden the scope and applicability of peroxidase-based proximity labeling.
Proximity-dependent biotinylation has revolutionized the study of protein-protein interactions and subcellular proteomics in living cells. This powerful methodology utilizes engineered enzymes, typically derived from biotin ligases, which are fused to a protein of interest to covalently tag nearby interacting and neighboring proteins with biotin. These biotinylated proteins can then be affinity-purified using streptavidin beads and identified via mass spectrometry, enabling the mapping of protein interaction networks within their native cellular environment.
The development of this technique addresses critical limitations of traditional methods for studying protein-protein interactions, such as co-immunoprecipitation and yeast two-hybrid systems. These conventional approaches often struggle to capture weak or transient interactions, are limited by antibody availability and specificity, and may not reflect the native physiological context of interactions within intact cells. Proximity labeling overcomes these hurdles by enabling the capture of interactions in living cells with high temporal resolution and the ability to target specific subcellular compartments.
This guide provides a comprehensive objective comparison between two primary tools in the proximity labeling toolkit: the established BioID method and the enhanced TurboID technology, focusing on their catalytic efficiencies, experimental parameters, and practical applications for researchers and drug development professionals.
Technical Principles and Evolutionary Development
BioID: The First-Generation Proximity Labeling Tool
BioID represents the foundational technology in proximity-dependent biotinylation. It utilizes a mutated form of the Escherichia coli biotin ligase (BirA), where an arginine-to-glycine substitution at position 118 (R118G) creates a promiscuous enzyme. This mutation substantially decreases the enzyme's affinity for its reaction intermediate, biotin-adenosine monophosphate (biotin-AMP), by approximately 440-fold compared to the wild-type enzyme [12] [13]. Consequently, instead of specifically transferring biotin to its normal target protein, the mutant enzyme (BirA*) releases the reactive biotin-AMP intermediate into the surrounding cellular environment, where it can covalently attach to lysine residues on nearby proteins within an estimated radius of 10-15 nm [14] [12].
A smaller variant, BioID2, was subsequently developed from the biotin ligase of Aquifex aeolicus. While BioID2 offers a reduced size that may minimize steric interference with the bait protein, it largely retains the catalytic characteristics of the original BioID, including similar temperature requirements and labeling kinetics [14] [15].
TurboID: Engineered for Enhanced Catalytic Performance
TurboID was developed through yeast display-based directed evolution of the BirA R118S mutant to address several limitations of the original BioID system [14]. This engineering process resulted in a biotin ligase with dramatically improved catalytic activity. The enhanced enzyme accelerates the conversion of biotin to the reactive biotin-AMP intermediate and increases the release rate of this intermediate, thereby significantly boosting the efficiency of proximal protein biotinylation [15].
A smaller version, miniTurbo, was also developed by removing the N-terminus of TurboID while maintaining its high catalytic activity. Both TurboID and miniTurbo represent substantial advancements in proximity labeling technology, offering researchers tools for rapid interaction capture in dynamic cellular environments [12] [13].
Comparative Mechanism of BioID and TurboID
Comparative Performance Analysis
Quantitative Performance Metrics
Extensive comparative studies across multiple biological systems have quantified the performance differences between BioID/BioID2 and TurboID. The data reveal substantial advantages for TurboID in several critical parameters essential for experimental design and implementation.
Table 1: Direct Performance Comparison of BioID2 and TurboID
| Performance Parameter | BioID2 | TurboID | Experimental Context |
|---|---|---|---|
| Minimum Labeling Time | 16-24 hours | 10 minutes - 3 hours | Xenopus embryos, mammalian cell lines [14] [12] |
| Optimal Temperature | 37°C+ | 13.6-24°C (room temperature compatible) | Xenopus embryo development [14] |
| Biotin Concentration | Requires exogenous biotin (50-500 μM) | Functions without exogenous biotin | Xenopus embryo culture [14] |
| Self-Biotinylation | Not detectable above background | Detectable even without biotin supplementation | Immunoblot with streptavidin-HRP [14] |
| Endogenous Background | Lower basal biotinylation | Higher background due to intense activity | Mammalian cell lines [12] [13] |
| Protein Stability | Generally stable | Signs of instability in some contexts | Stable cell lines [12] |
The dramatically reduced labeling time required by TurboID (as little as 10 minutes under optimal conditions) compared to BioID2 (typically 16-24 hours) enables researchers to capture rapid biological processes, such as transient signaling events, rapid cellular responses to stimuli, and dynamic protein complex assembly and disassembly [14] [12]. This enhanced temporal resolution is particularly valuable for studying processes that occur on minute-to-hour timescales rather than day-long timescales.
Temperature Tolerance and Application Range
Traditional BioID enzymes function optimally at 37°C or higher, which restricts their application in temperature-sensitive systems. BioID2 demonstrates maximal efficiency at even higher temperatures around 50°C, further limiting its practical utility [14]. In contrast, TurboID maintains significant activity across a broad temperature range (13.6-24°C), making it compatible with diverse biological systems including developing embryos, plant tissues, and other temperature-sensitive models that cannot tolerate elevated temperatures [14] [15].
This temperature flexibility was convincingly demonstrated in Xenopus embryos, where TurboID effectively biotinylated proximal proteins at all tested temperatures compatible with normal embryonic development, while BioID2 showed no detectable activity under the same conditions [14]. This significantly expands the potential applications of proximity labeling to previously challenging experimental systems.
Biotin Requirements and Cellular Toxicity
A notable operational difference between these systems concerns their biotin requirements. BioID2 typically requires supplementation with relatively high concentrations of exogenous biotin (50-500 μM) to achieve detectable labeling, whereas TurboID can function effectively with endogenous cellular biotin levels, demonstrating self-biotinylation even without supplementation [14]. This characteristic simplifies experimental design for TurboID applications.
However, TurboID's enhanced catalytic activity presents challenges related to cellular toxicity. High basal activity and consequent biotin depletion can adversely affect cell viability, particularly in sensitive systems [12] [16]. Recent methodological advancements address this limitation through biotin-blockage protocols, where free biotin levels are controlled using biotin scavengers until labeling is intentionally initiated with exogenous biotin supplementation [16]. This approach maintains inducibility while mitigating toxicity concerns, expanding TurboID's applicability in proteomic studies.
Experimental Design and Methodologies
Standardized Experimental Workflow
Proximity Labeling Experimental Flow
Critical Experimental Considerations
Control Design
Appropriate controls are essential for generating high-confidence interactome data. Recent studies demonstrate that background proteins in proximity labeling experiments arise from both bead adsorption and self-labeling by the biotin ligase itself [17]. Expression-matched controls, where control samples express TurboID or TurboID-GFP at levels precisely matching the bait fusion protein, significantly reduce background interference and improve interactome assignment accuracy [17]. Discordant expression levels between bait and control samples frequently lead to both false-positive and false-negative identifications.
Biotin Concentration Optimization
For TurboID experiments, biotin concentration requires careful optimization to balance labeling efficiency against potential cellular toxicity. While TurboID can function with endogenous biotin levels, supplementation with 50-500 μM biotin typically enhances labeling intensity [14] [12]. For inducible systems, biotin-blockage protocols using commercial biotin scavengers can control basal biotinylation activity until labeling is intentionally initiated with exogenous biotin [16].
Subcellular Localization Validation
Verifying correct subcellular localization of the bait-TurboID/BioID fusion protein is critical before proceeding with large-scale experiments. Immunofluorescence staining with tags such as HA or Myc enables confirmation that the fusion protein localizes appropriately and maintains proper function [12] [13]. Mislocalized fusion proteins will generate irrelevant interactome data regardless of labeling efficiency.
Specialized Protocol: TurboID in Plant Systems
TurboID has been successfully adapted for plant systems, where it outperforms BioID due to its room temperature activity. A standardized protocol for Nicotiana benthamiana includes these key steps [15]:
Agroinfiltration: Agrobacterium tumefaciens strain GV3101 carrying TurboID constructs (p35S:Citrine-TurboID-3xHA or pUBQ:Citrine-TurboID-3xMyc) are infiltrated into plant leaves.
Biotin Treatment: Infiltrated tissues are treated with 50 mM biotin dissolved in 100 mM sodium phosphate buffer (pH 7.2) for specified durations.
Protein Extraction: Tissues are ground in liquid nitrogen and proteins extracted using appropriate lysis buffers.
Biotin Removal: Free biotin is removed using desalting columns to reduce background in downstream applications.
Affinity Purification: Biotinylated proteins are captured using streptavidin-coated beads under stringent washing conditions.
This protocol has been successfully applied to study immune receptor interactions in plants, demonstrating TurboID's versatility across diverse biological systems [15].
Research Reagent Solutions
Table 2: Essential Research Reagents for Proximity Labeling Studies
| Reagent Category | Specific Examples | Function and Application Notes |
|---|---|---|
| Expression Plasmids | 3xHA-TurboID (Addgene #107171), 3xHA-miniTurbo (Addgene #107172), mycBioID pBabe (Addgene #80901) | Critical to select appropriate fusion configuration for bait protein; N-terminal vs C-terminal fusions may affect protein function [12] [13] |
| Cell Culture Reagents | Dialyzed FBS, Biotin (50 mM stock), Doxycycline (for inducible systems) | Dialyzed serum reduces background biotin; biotin concentration must be optimized for specific system [12] [16] |
| Affinity Purification Materials | Streptavidin-coated beads, Magnetic bead systems | High-quality streptavidin beads essential for reducing non-specific binding; magnetic systems simplify processing [15] [17] |
| Detection Reagents | Streptavidin-HRP, Anti-HA antibodies, Fluorescent streptavidin conjugates | Multiple detection methods allow validation at different stages; streptavidin-HRP for immunoblot, fluorescent conjugates for microscopy [14] [12] |
| Specialized Reagents | Biotin scavengers (e.g., BioBlock), Protease inhibitor cocktails | Biotin scavengers enable inducible TurboID systems; protease inhibitors maintain protein integrity during extraction [16] |
Applications Across Biological Systems
TurboID's enhanced catalytic efficiency has expanded applications of proximity labeling to previously challenging biological contexts:
Neuroscience Applications
In neuroscience, TurboID has enabled mapping of protein interaction networks within specific neuronal compartments and cell types. Its rapid labeling kinetics are particularly valuable for capturing dynamic processes such as synaptic remodeling, calcium-dependent signaling, and neurotransmitter receptor trafficking [18]. TurboID has been applied to identify protein interactions altered in neurological disorders including autism, schizophrenia, and neurodegenerative diseases, providing insights into disease mechanisms and potential therapeutic targets [18].
Plant Biology Applications
Traditional BioID approaches have limited utility in plant systems due to their suboptimal temperature requirements and extended labeling times. TurboID functions effectively at room temperature and with shorter labeling durations, enabling proximity labeling studies in Arabidopsis, Nicotiana benthamiana, and other plant species [15]. This has facilitated the identification of interactors for plant immune receptors, components of signaling pathways, and proteins involved in development and stress responses [15].
Developmental Biology Studies
The rapid developmental processes in model systems such as Xenopus embryos and zebrafish benefit significantly from TurboID's faster labeling capabilities. Early embryonic divisions occurring in 20-30 minute intervals can be captured with TurboID but would be missed with traditional BioID approaches requiring 16+ hour labeling periods [14]. This temporal resolution enables the mapping of protein interactome dynamics during critical developmental transitions.
Emerging Innovations and Future Perspectives
The field of proximity labeling continues to evolve with several promising developments building upon the TurboID platform:
Split-TurboID Systems
Split-TurboID separates the enzyme into two inactive fragments that reconstitute functionality only when brought together through specific protein-protein interactions [18]. This approach increases specificity by labeling proteins proximal to interaction interfaces rather than all proteins near the bait. While technically more challenging, this system provides higher resolution mapping of direct interaction networks.
Environment-Activated Labeling Systems
Recent innovations include biotin ligases engineered to respond to specific cellular conditions. For example, Cal-ID incorporates calmodulin to create a calcium-sensitive biotin ligase that activates upon local calcium ion fluctuations, enabling biochemical recording of calcium signaling events in neurons [19]. Similar approaches are being developed for other signaling molecules and cellular conditions.
Endogenous Tagging Strategies
While most proximity labeling studies rely on overexpression of fusion proteins, recent advances facilitate tagging of endogenous loci using CRISPR/Cas9 systems or antibody-enzyme conjugates that target native proteins without genetic manipulation [19]. These approaches minimize artifacts associated with overexpression and enable studies in primary cells and tissues where genetic manipulation is challenging.
The evolution from BioID to TurboID represents a significant advancement in proximity-dependent biotinylation technology, primarily through dramatic improvements in catalytic efficiency. TurboID's rapid labeling kinetics, temperature flexibility, and reduced biotin requirements make it superior for studying dynamic cellular processes and applying proximity labeling to previously challenging biological systems.
However, BioID retains utility for applications where slower, more controlled labeling is desirable or where TurboID's high activity causes excessive background or cellular toxicity. The choice between these systems should be guided by specific experimental requirements, including temporal resolution needed, biological system constraints, and tolerance for potential background labeling.
As the proximity labeling toolkit continues to expand with increasingly specialized enzymes and methodologies, researchers have unprecedented capability to map protein interaction networks with high spatiotemporal resolution in living systems. These advances promise to deepen our understanding of cellular organization and function in both health and disease.
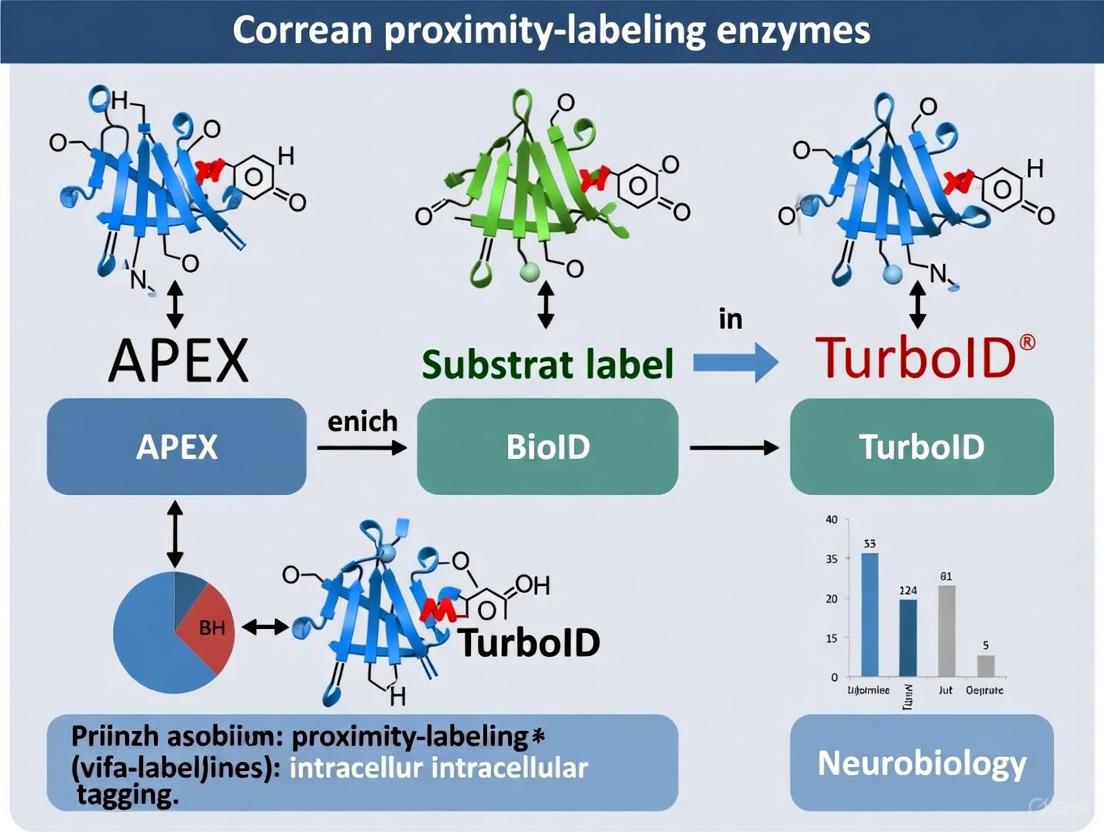
The evolution of proximity-labeling enzymes represents a transformative journey in molecular biology, enabling researchers to capture intricate protein-protein interactions and dynamic cellular processes within their native environments. This field has progressed from the foundational, naturally occurring biotin ligase BirA to a suite of engineered enzymes with enhanced capabilities, largely driven by the powerful method of directed evolution. Directed evolution mimics natural selection in the laboratory through iterative rounds of genetic diversification, screening, and amplification of biological entities with desired traits [20]. For proximity labeling, this process has been instrumental in overcoming the limitations of initial tools, such as slow labeling kinetics and dependence on exogenous co-factors, leading to the development of high-performance enzymes like TurboID and split-TurboID that are revolutionizing intracellular tagging research [21] [18]. This guide objectively compares the performance of these key enzymes, providing the experimental data and methodologies essential for researchers selecting the optimal tool for their specific applications.
Foundational Technology: The Emergence of BirA
The cornerstone of biotin-based proximity labeling is the Escherichia coli enzyme BirA. In its wild-type form, BirA is a highly specific biotin protein ligase that catalyzes the covalent attachment of biotin to a single lysine residue on its endogenous substrate, the biotin carboxyl carrier protein (BCCP) subunit of acetyl-CoA carboxylase [21] [22]. The catalytic mechanism involves BirA using ATP to convert biotin into a reactive intermediate, biotinoyl-5'-AMP. In its wild-type state, BirA retains this intermediate tightly within its active site until the specific acceptor protein or peptide is encountered [21].
The critical breakthrough came with the mutation of a key residue in the BirA active site (R118G), which reduced the enzyme's affinity for biotinoyl-5'-AMP. This mutated version, known as BioID, promiscuously releases the reactive biotin intermediate, allowing it to covalently tag lysine residues on any proximal proteins within an estimated 10 nm radius [21]. This innovation transformed BirA from a specific metabolic enzyme into a general-purpose proximity-labeling tool.
Table 1: Key Characteristics of Foundational Proximity-Labeling Enzymes
| Enzyme | Origin | Key Mutations/Features | Primary Labeling Substrate | Typical Labeling Time | Estimated Labeling Radius |
|---|---|---|---|---|---|
| BirA (Wild-Type) | E. coli | None (High-specificity) | Biotinoyl-5'-AMP | N/A (Specific to BCCP) | N/A |
| BioID | E. coli | R118G | Biotinoyl-5'-AMP | 18-24 hours | ~10 nm |
| BioID2 | Aquifex aeolicus | R40G, lacks N-terminal DNA-binding domain | Biotinoyl-5'-AMP | ~18 hours | ~10 nm |
| APEX2 | Soybean (Ascorbate Peroxidase) | K14D, E112K, and other enhancements | Biotin-phenol radicals | <1 minute | <20 nm |
However, the first-generation BioID had significant limitations for dynamic cellular studies. Its slow labeling kinetics, requiring 18-24 hours of biotin incubation, made it unsuitable for capturing transient interactions or mapping rapid biological processes [21] [18]. This limitation, coupled with its relatively large size which could sterically hinder the bait protein, set the stage for the next phase of innovation through directed evolution.
The Directed Evolution Revolution
Directed evolution is an iterative laboratory process that harnesses the principles of natural selection—genetic diversification, selection for desired traits, and amplification—to steer biomolecules toward user-defined goals [20] [23]. The process does not require prior structural knowledge, allowing for the improvement of protein functions even when the effects of mutations are difficult to predict [23].
The general workflow involves:
- Diversification: Creating a large library of gene variants through random mutagenesis (e.g., error-prone PCR) or recombination-based methods (e.g., DNA shuffling) [20] [24].
- Selection/Screening: Employing a high-throughput assay to identify the rare variants with improved properties. This can be a selection, where activity is coupled to survival, or a screening system where each variant is quantitatively assayed [23].
- Amplification: The genes of the best-performing variants are isolated and amplified to serve as templates for the next round of evolution [20].
This methodology has been successfully applied to evolve numerous enzymes for industrial and research applications, including the enhancement of biotin ligases for proximity labeling [20] [25]. For example, one study utilized in vitro compartmentalization to evolve BirA variants with altered substrate specificity towards the biotin analog desthiobiotin [25]. Another employed a bacterial display system to efficiently select for novel BirA variants capable of biotinylating peptide sequences from unmodified proteins [22].
Engineered Enzymes and Performance Comparison
The application of directed evolution to BioID led to a quantum leap in performance with the development of TurboID and miniTurbo. These enzymes were evolved via yeast display screening of a mutagenized BioID library for increased biotinylation activity [21]. TurboID dramatically accelerates labeling times from hours to minutes, achieving in 10 minutes an equivalent level of biotinylation that BioID requires 18 hours to accomplish [21] [18]. While TurboID's enhanced activity is a major advantage, it can lead to increased background labeling and potential cellular toxicity if not carefully optimized by controlling labeling time and biotin concentration [18].
A further innovation is split-TurboID, where the enzyme is separated into two inactive fragments. Labeling only occurs when the fragments are reconstituted through a specific protein-protein interaction, enabling highly precise mapping of interactomes [18]. This system has been ingeniously adapted to sense other cellular events, such as calcium influx. The Ca2+-activated split-TurboID (CaST) system tethers the split fragments to Ca2+/calmodulin domains, causing them to reconstitute and label proteins only in the presence of elevated intracellular calcium, thus acting as a rapid, non-invasive reporter of neuronal activity [26].
Table 2: Performance Comparison of Evolved Proximity-Labeling Enzymes
| Enzyme | Size (Amino Acids) | Catalytic Rate | Optimal Labeling Time | Key Advantages | Key Limitations |
|---|---|---|---|---|---|
| TurboID | ~35 kDa (Full length) | Very High | 10 - 30 minutes | Ultra-fast labeling, high sensitivity for transient interactions | Potential cellular toxicity, higher background |
| miniTurbo | ~28 kDa | High | 30 minutes - 2 hours | Smaller size, reduced background without biotin | Lower activity than TurboID |
| split-TurboID | N/A (Two fragments) | Activated by Reconstitution | Varies with reconstitution | High specificity for protein complexes and cellular events | Requires reconstitution, more complex experimental setup |
| APEX2 | ~27 kDa | Very High (Limited by H₂O₂) | <1 minute | Ultrafast, compatible with EM | Requires H₂O₂, potential oxidative stress |
Directed Evolution Workflow for TurboID
Experimental Protocols and Data Analysis
Protocol for Ca2+-Activated Split-TurboID (CaST) in Neuronal Tagging
The CaST system exemplifies a sophisticated application of evolved proximity labeling. The following methodology is adapted from its use for tagging prefrontal cortex neurons activated by psilocybin in untethered mice [26].
- Molecular Constructs: The two components of CaST are typically encoded in a single bicistronic vector (e.g., CaST-IRES) to ensure coordinated expression in the same cell. The optimized construct uses a membrane-tethered CD4-sTb(C)-M13-GFP and a cytosolic CaM-V5-sTb(N) [26].
- Transfection/Transduction: Introduce the CaST construct into the target cells (e.g., HEK293T for validation, or neurons via AAV delivery in vivo).
- Activity Labeling:
- Prepare a solution of biotin in an appropriate buffer (e.g., artificial cerebrospinal fluid for in vivo work). Biotin concentration must be optimized, but is typically in the low millimolar range.
- Deliver the biotin solution to the cells or animal systemically (e.g., intraperitoneal injection). The labeling window is user-defined and can be as short as 10 minutes [26].
- Tissue Processing and Readout:
- Immediately after the labeling period, euthanize the animal and perfuse to remove excess biotin.
- Dissect the brain region of interest and prepare tissue for either:
- Immunohistochemistry: Fix tissue, section, and incubate with fluorescently conjugated Streptavidin (e.g., SA-647) and other desired antibodies to visualize biotinylated proteins [26].
- Western Blot: Lyse tissue, run SDS-PAGE, and probe with streptavidin-HRP to confirm bulk biotinylation [26].
- Mass Spectrometry: Enrich biotinylated proteins using streptavidin beads, followed by on-bead tryptic digest and LC-MS/MS for proteomic identification.
Advanced Proteomic Analysis: From Protein- to Peptide-Level Enrichment
Conventional analysis involves enriching entire biotinylated proteins on streptavidin beads before digestion and MS analysis. However, a novel "super-resolution proximity labeling" method significantly improves accuracy by enriching for biotinylated peptides specifically [27].
This advanced protocol involves:
- Precipitation and Digestion: Precipitate protein extracts to remove excess biotin, then denature and digest with trypsin.
- Peptide-Level Capture: Incubate the digested peptides (not intact proteins) with streptavidin beads to capture only the biotinylated peptides.
- Stringent Washing and Elution: Wash the beads stringently and elute the biotinylated peptides using an acidic organo-aqueous denaturation buffer.
- LC-MS/MS Analysis: The eluted peptides are directly analyzed by LC-MS/MS without further purification [27].
This method provides direct evidence of biotinylation sites, eliminates the need for negative controls for fold-change calculations, and reduces false positives by avoiding the co-enrichment of non-biotinylated peptides from labeled proteins. It achieved a ~89% True Positive Rate (TPR) for mitochondrial matrix proteins without a negative control, compared to a TPR of ~78.8% for the conventional approach [27].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagent Solutions for Proximity Labeling Experiments
| Reagent / Solution | Function / Role | Example Use Case |
|---|---|---|
| Biotin | Core substrate for BioID, TurboID, and split-enzymes. Converted to reactive biotin-AMP. | Essential for all biotin ligase-based labeling; concentration and time must be optimized [26] [21]. |
| Streptavidin-Conjugated Beads | High-affinity capture of biotinylated proteins for enrichment and purification. | Used in pull-down assays prior to mass spectrometric analysis or western blotting [21] [27]. |
| Fluorescently-Labeled Streptavidin (e.g., SA-647) | Visualizing the spatial distribution of biotinylation via microscopy. | Critical for immunohistochemical readout of labeling efficiency and pattern, as in CaST experiments [26]. |
| Desthiobiotin | A biotin analog used in some evolved BirA variants; allows for gentler elution from streptavidin due to lower affinity. | Can improve recovery of labeled proteins in pull-downs [25]. |
| Digoxigenin (DIG)-Modified Oligos & HyPro Enzyme | For RNA-proximity labeling (HyPro). Oligos target the HyPro enzyme (APEX2 derivative) to specific RNA molecules. | Enables mapping of the proteome associated with specific RNA transcripts or compartments [4]. |
| Trehalose-Based Labeling Buffer | Increases viscosity of the labeling reaction medium to limit diffusion of activated biotin radicals. | Enhances spatial resolution by reducing the spread of the label, crucial for small compartments [4]. |
The historical evolution from BirA to directedly evolved enzymes like TurboID and its derivatives has fundamentally expanded the toolbox for intracellular tagging research. The quantitative data and protocols presented here underscore a clear trajectory of innovation: each generation of tools offers significant gains in speed, specificity, and versatility. While first-generation BioID established the core principle, TurboID enabled the study of dynamic processes, and systems like split-TurboID and CaST now provide exquisite temporal and conditional control. The ongoing refinement of experimental protocols, particularly the shift towards peptide-level proteomic analysis, promises even greater accuracy and depth in mapping the intricate molecular interactions that define cellular function. For the researcher, this evolution means that selecting the right tool requires careful consideration of the biological question, balancing the need for speed against potential toxicity, and the requirement for specificity against experimental complexity.
Proximity labeling (PL) has emerged as a powerful technology for capturing biomolecular interactions in living systems, enabling researchers to gain new insights into protein-protein interactions, RNA-protein interactions, and cellular compartment proteomics. This technique involves using genetically engineered enzymes that generate reactive species to covalently tag neighboring molecules with biotin within a limited radius, allowing for subsequent affinity purification and identification. As the PL field has rapidly advanced, researchers now have access to a diverse toolkit of enzymes with varying characteristics, performance parameters, and applications. This comparison guide provides an objective analysis of the key enzymatic tools for intracellular tagging research, focusing on their defining characteristics: labeling radius, kinetics, and residue specificity, to inform selection for specific experimental needs.
Enzyme Systems and Mechanisms
Proximity labeling enzymes can be broadly categorized into two main classes: peroxidases and biotin ligases, each with distinct catalytic mechanisms and operational requirements.
Figure 1: Classification of major proximity labeling enzyme systems with their key representatives.
Peroxidase-based systems such as APEX, APEX2, and HRP utilize hydrogen peroxide (H₂O₂) to oxidize biotin-phenol substrates into phenoxyl radicals that covalently attach to electron-rich amino acids on nearby proteins [28] [8] [29]. These radicals have an extremely short lifespan (<1 ms) and limited diffusion capability, resulting in a tight labeling radius approximately <20 nm from the enzyme [28] [8]. The key advantage of peroxidase systems is their rapid labeling capability, with reactions typically complete within 1 minute, enabling precise temporal control [28] [29]. However, a significant limitation is the requirement for H₂O₂, which can be toxic to cells and complicate in vivo applications [28] [29].
Biotin ligase-based systems, including BioID, TurboID, and miniTurbo, operate through a distinct mechanism where the enzyme utilizes ATP to activate biotin into biotin-5'-AMP, which then covalently attaches to lysine residues on proximal proteins [28] [30] [29]. These systems offer the advantage of not requiring H₂O₂, making them more suitable for in vivo applications [28]. However, they typically have slower kinetics, with labeling times ranging from 10 minutes to 24 hours depending on the specific enzyme variant [28]. The labeling radius for biotin ligases has been estimated at approximately 10 nm, though this may vary based on the specific enzyme, subcellular compartment, and labeling duration [28].
Recent advancements have introduced specialized and engineered systems that address specific research needs. Split-enzyme systems like Split-TurboID and CaST (Ca²⁺-activated split-TurboID) provide activity-dependent labeling capabilities [6]. Environment-activated enzymes such as Cal-ID respond to calcium fluctuations [19], while light-activated systems including LITag and LOV-TurboID enable spatiotemporal control through blue light illumination [19]. Newer H₂O₂-independent enzymes like BmTyr (bacterial tyrosinase) and LaccID utilize molecular oxygen instead of H₂O₂, offering improved biocompatibility [19].
Comparative Performance Analysis
The selection of an appropriate proximity labeling enzyme requires careful consideration of multiple performance parameters tailored to specific experimental requirements and biological contexts.
Table 1: Comprehensive Comparison of Proximity Labeling Enzyme Characteristics
| Enzyme | Labeling Radius | Kinetics (Labeling Time) | Residue Specificity | Key Advantages | Major Limitations |
|---|---|---|---|---|---|
| APEX/APEX2 | <20 nm [28] [8] | ~1 min [28] [29] | Tyr, Trp, His, Cys [28] | Rapid labeling, excellent temporal control | H₂O₂ toxicity, not ideal for in vivo |
| HRP | <20 nm [28] | ~1 min [28] | Tyr, Trp, His, Cys [28] | Well-characterized, multiple commercial substrates | Limited to secretory pathway/oxidizing environments |
| BioID | ~10 nm [28] | 15-24 h [28] [29] | Lys, protein N-termini [28] | Works in vivo, minimal toxicity | Very slow kinetics, poor temporal control |
| TurboID | ≥35 nm (time-dependent) [28] | 10 min [28] [29] | Lys, protein N-termini [28] | Rapid labeling, works in vivo | Cellular toxicity at high expression, background labeling |
| miniTurbo | ~10 nm [28] | 10 min-1 h [29] | Lys, protein N-termini [28] | Rapid labeling, less background than TurboID | Lower activity than TurboID |
| BmTyr | Not specified | ≤10 min [19] | Tyr-specific [19] | H₂O₂-free, improved biocompatibility | Efficiency may decrease in hypoxic conditions |
| LaccID | Not specified | 1-2 h [19] | Presumably electron-rich residues | H₂O₂-free, uses O₂ | Limited to cell surface applications |
| CaST | Not specified | <10 min [6] | Lys, protein N-termini [6] | Calcium-activated, temporal control | Requires biotin supplementation |
The labeling radius represents a critical parameter that determines spatial resolution. Peroxidase-based systems generally offer superior spatial resolution due to the extremely short-lived nature of the phenoxyl radicals they generate [28] [8]. In contrast, biotin ligase-based systems exhibit more variable labeling radii, with TurboID demonstrating a notably larger radius (≥35 nm) that increases with longer labeling times [28]. This expanded radius may be advantageous for capturing broader interactomes but reduces spatial precision.
Kinetic parameters directly impact temporal resolution and experimental design. Peroxidase systems provide the fastest labeling capabilities, with reactions typically complete within 1 minute, enabling near-instantaneous snapshotting of interactomes [28] [29]. Biotin ligases show considerable variation in their kinetics, with traditional BioID requiring 15-24 hours while engineered variants like TurboID and miniTurbo achieve labeling in as little as 10 minutes [28] [29]. The recent development of BmTyr further bridges this gap with ≤10 minute labeling without H₂O₂ requirements [19].
Residue specificity determines the potential labeling space and efficiency within the proteome. Peroxidases preferentially target electron-rich amino acids including tyrosine, tryptophan, histidine, and cysteine [28]. Biotin ligases exclusively target lysine residues and protein N-termini [28]. This fundamental difference means that the same protein may present different labeling profiles when analyzed with different enzyme systems, depending on the surface accessibility of these target residues.
Experimental Protocols and Methodologies
Implementing proximity labeling requires careful experimental design and optimization. Below are detailed protocols for key enzyme systems based on established methodologies.
APEX2 Labeling Protocol for Subcellular Proteomics
This protocol outlines the standard procedure for APEX2-mediated proximity labeling to capture subcellular proteomes, adapted from established methodologies [8].
Reagents and Solutions:
- Biotin-phenol stock solution (500 mM in DMSO)
- H₂O₂ working solution (1 mM prepared fresh in PBS)
- Quenching solution (TROLOX, sodium ascorbate, and sodium azide in PBS)
- Lysis buffer (50 mM Tris-HCl, pH 7.4, 500 mM NaCl, 0.2% SDS, 1% Triton X-100, protease inhibitors)
- Streptavidin-coated magnetic beads
Procedure:
- Cell Preparation: Culture cells expressing APEX2 fusion protein and include untransfected controls. Grow to 70-90% confluency.
- Biotin-phenol Incubation: Add biotin-phenol to culture medium at final concentration of 500 μM. Incubate for 30 minutes at 37°C.
- H₂O₂ Activation: Add H₂O₂ to final concentration of 1 mM. Incubate for exactly 1 minute at room temperature.
- Quenching: Immediately remove H₂O₂-containing medium and add ice-cold quenching solution. Wash cells three times with quenching solution.
- Cell Lysis: Harvest cells using lysis buffer. Incubate on ice for 30 minutes with occasional vortexing.
- Clarification: Centrifuge lysates at 15,000 × g for 15 minutes at 4°C.
- Streptavidin Enrichment: Incubate clarified lysate with streptavidin-coated magnetic beads for 1 hour at room temperature.
- Washing: Wash beads sequentially with:
- Lysis buffer (2 times)
- High-salt wash buffer (1 M KCl, 0.1% Na₂CO₃)
- Urea wash buffer (2 M urea in 10 mM Tris-HCl, pH 8.0)
- RIPA buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate)
- On-bead Digestion: Add trypsin in 50 mM Tris-HCl, pH 8.0 and digest overnight at 37°C.
- Mass Spectrometry Analysis: Desalt peptides and analyze by LC-MS/MS.
TurboID Labeling Protocol for In Vivo Applications
This protocol describes TurboID-mediated labeling suitable for live cells and in vivo applications, based on established methods [28] [29].
Reagents and Solutions:
- Biotin stock solution (50 mM in DMSO)
- Lysis buffer (50 mM Tris-HCl, pH 7.4, 500 mM NaCl, 0.2% SDS, 1% Triton X-100, protease inhibitors)
- Streptavidin-coated magnetic beads
Procedure:
- Cell Preparation: Culture cells expressing TurboID fusion protein.
- Biotin Incubation: Add biotin to culture medium at final concentration of 500 μM. For in vivo applications, administer biotin via intraperitoneal injection (50 mg/kg).
- Labeling Duration: Incubate for desired time (10 minutes to several hours depending on experimental needs).
- Cell Harvesting: Remove medium and wash cells with PBS.
- Cell Lysis: Harvest cells using lysis buffer. Incubate on ice for 30 minutes with occasional vortexing.
- Clarification: Centrifuge lysates at 15,000 × g for 15 minutes at 4°C.
- Streptavidin Enrichment: Incubate clarified lysate with streptavidin-coated magnetic beads for 1 hour at room temperature.
- Washing: Wash beads sequentially with lysis buffer, high-salt buffer, and RIPA buffer.
- Elution: Elute biotinylated proteins with Laemmli buffer containing 2 mM biotin and 20 mM DTT at 95°C for 10 minutes, or proceed to on-bead digestion for mass spectrometry.
Enhanced HyPro Technology for RNA-Protein Interactions
This specialized protocol describes the enhanced hybridization-proximity labeling (HyPro) technology for mapping protein interactomes of single RNA molecules, based on recent methodological advances [4].
Reagents and Solutions:
- Modified HyPro2 enzyme
- Digoxigenin (DIG)-modified antisense oligonucleotides
- Biotin-tyramide working solution
- H₂O₂ working solution
- Trehalose-containing labeling buffer
- Streptavidin affinity purification reagents
Procedure:
- Cell Fixation and Permeabilization: Fix cells with 4% formaldehyde for 10 minutes, then permeabilize with 0.5% Triton X-100 for 5 minutes.
- Hybridization: Incubate with DIG-modified antisense oligonucleotides targeting RNA of interest in hybridization buffer overnight at 37°C.
- HyPro Enzyme Binding: Incubate with modified HyPro2 enzyme (2 μg/mL) in trehalose-containing labeling buffer for 30 minutes at room temperature.
- Proximity Labeling: Add biotin-tyramide to final concentration of 500 μM and H₂O₂ to 1 mM. Incubate for exactly 1 minute.
- Quenching and Washes: Quench with TROLOX/sodium ascorbate solution and wash extensively.
- Streptavidin Affinity Purification: Lyse cells and incubate with streptavidin magnetic beads for 1 hour.
- Proteomic Analysis: Process enriched proteins for mass spectrometry analysis.
Figure 2: Generalized workflow for proximity labeling experiments showing key decision points and protocol options.
Research Reagent Solutions
Successful implementation of proximity labeling experiments requires specific reagents and materials optimized for each enzyme system.
Table 2: Essential Research Reagents for Proximity Labeling Experiments
| Reagent Category | Specific Examples | Function and Application Notes |
|---|---|---|
| Enzyme Systems | APEX2, TurboID, miniTurbo, HyPro2 | Core labeling enzymes; selection depends on required kinetics, resolution, and cellular context |
| Activation Reagents | H₂O₂ (for peroxidases), Biotin (for ligases), Blue light (for LOV-TurboID) | Enzyme-specific activators; concentration and timing critically impact labeling efficiency and specificity |
| Labeling Substrates | Biotin-phenol (for peroxidases), Biotin (for ligases) | Precursors for reactive labeling species; membrane permeability varies |
| Quenching Solutions | TROLOX, sodium ascorbate, sodium azide | Terminate labeling reactions; essential for temporal control |
| Affinity Matrices | Streptavidin-coated magnetic beads | Capture biotinylated proteins; bead size and coating density affect enrichment efficiency |
| Lysis Buffers | RIPA, SDS-containing buffers, Trehalose buffers | Extract labeled proteins while maintaining integrity; trehalose reduces diffusion artifacts |
| Wash Solutions | High-salt buffers, urea-containing buffers, detergent solutions | Remove nonspecifically bound proteins; stringency affects background |
| Elution Reagents | Laemmli buffer with biotin and DTT, On-bead digestion | Release captured proteins for analysis; method depends on downstream application |
Advanced Applications and Emerging Technologies
The proximity labeling field continues to evolve with novel enzymes and applications expanding the technology's capabilities.
Environment-Activated Systems: Recent innovations include enzymes that respond to specific physiological conditions. Cal-ID represents an engineered biotin ligase that senses local calcium fluctuations through calmodulin, enabling spatially resolved biochemical recording of Ca²⁺ signaling and neuronal activity [19]. Similarly, ROS-activated PL systems leverage endogenous reactive oxygen species as a source of H₂O₂ to activate APEX-mediated labeling, allowing monitoring of oxidative events specifically within ROS hotspots [19].
RNA-Centric Applications: Enhanced HyPro technology demonstrates the adaptation of PL for challenging targets including single RNA molecules. By re-engineering the HyPro enzyme and optimizing proximity biotinylation conditions, researchers can now identify proteins associated with compact RNA-containing nuclear bodies, small pre-mRNA clusters, and individual transcripts [4]. This approach has revealed extensive interactions between pathogenic G4C2 repeat-containing C9orf72 RNAs and disease-linked paraspeckle markers in ALS patient-derived cells [4].
Endogenous Targeting Strategies: Moving beyond genetically encoded fusion proteins, recent approaches enable PL for endogenous targets through ligand-directed and antibody-directed strategies. These include aptamer-HRP conjugates for selective modification of target cells [19], small molecule ligand-enzyme fusions for mapping proximal proteomes of endogenous neurotransmitter receptors [19], and antibody-photocatalyst conjugates for mapping interactomes of native proteins in unmodified biological systems [19].
Cascade Reaction Systems: Multi-enzyme PL systems enhance spatial selectivity through engineered biological cascades. For example, combining singlet oxygen photosensitizing protein with APEX2 enables H₂O₂ generation followed by proximity labeling without exogenous H₂O₂ addition [19]. Similarly, the two-level spatially localized proximity labeling (P2L) system incorporates galactose oxidase to generate H₂O₂ from specific glycans prior to the HRP-mediated labeling step [19].
Proximity labeling technologies have revolutionized our ability to capture biomolecular interactions in living systems, with enzyme characteristics fundamentally determining experimental outcomes. The labeling radius, kinetics, and residue specificity of each enzyme system establish its optimal applications—from high-resolution spatial mapping with peroxidase-based systems to in vivo interaction capture with advanced biotin ligases. As the field progresses toward more sensitive, specific, and physiologically compatible tools, researchers must carefully match enzyme characteristics to biological questions. The continued development of condition-activated, RNA-targeted, and endogenous-specific labeling systems promises to further expand the applications of these powerful technologies in basic research and drug development.
From Tool to Insight: Strategic Application of PL Enzymes in Biological Research
Proximity-dependent labeling (PL) has revolutionized the study of spatial proteomics and molecular interactions in living cells. This guide objectively compares the performance of major PL enzymes—including BioID, APEX, TurboID, and their variants—across different model systems, supported by experimental data.
Enzyme-catalyzed proximity labeling (PL) has emerged as a powerful alternative to traditional methods like affinity purification and yeast two-hybrid systems for mapping protein-protein interactions, organelle proteomes, and protein-nucleic acid interactions [31] [8]. These techniques utilize engineered enzymes that generate reactive molecules to covalently tag neighboring proteins and other biomolecules within a limited radius in live cells [28] [21]. The tagged molecules can then be isolated using streptavidin-based purification and identified via mass spectrometry [32]. A key advantage of PL is its ability to capture weak, transient interactions and insoluble proteins that are difficult to study with traditional methods, all while maintaining cellular physiological conditions [29].
Comparative Analysis of Proximity Labeling Enzymes
PL enzymes primarily fall into two categories: peroxidases (e.g., APEX/APEX2) and biotin ligases (e.g., BioID, TurboID) [28] [8]. The table below summarizes their key characteristics and performance metrics.
Table 1: Comparison of Major Proximity Labeling Enzymes
| Enzyme | Class | Size (kD) | Labeling Time | Labeling Radius | Primary Substrate | Key Advantages | Major Limitations |
|---|---|---|---|---|---|---|---|
| BioID [3] [29] | Biotin Ligase | ~35 | 18-24 hours [3] | ~10 nm [21] | Biotin | Low background; well-established | Very slow kinetics |
| BioID2 [29] | Biotin Ligase | ~27 | >16 hours [3] | ~10 nm | Biotin | Smaller size; less biotin required | Still requires long labeling times |
| TurboID [3] [29] | Biotin Ligase | 35 | 10 minutes [3] | ≥35 nm [28] | Biotin | Extremely fast; high signal | Some toxicity; baseline activity |
| miniTurbo [3] [29] | Biotin Ligase | 28 | 10 minutes [3] | Similar to TurboID [28] | Biotin | Fast; lower background than TurboID | ~2x less active than TurboID [3] |
| APEX/APEX2 [28] [29] | Peroxidase | ~28 | 1 minute [28] | <20 nm [8] | Biotin-phenol + H₂O₂ | Fastest; works for EM [8] | H₂O₂ toxicity; poor membrane permeability of substrate |
Performance Data from Key Experiments
Direct comparative experiments highlight the significant performance differences between these enzymes. In a foundational study, when expressed in the cytosol of HEK 293T cells, TurboID and miniTurbo biotinylated endogenous proteins much more rapidly than BioID, showing a ~3-6-fold difference in signal at early time points and a ~15-23-fold difference at later time points [3]. TurboID produced as much biotinylated product in 10 minutes as BioID, BioID2, or BASU generated in 18 hours [3].
Furthermore, the performance of these enzymes is context-dependent. A comparison of TurboID, miniTurbo, and BioID across different cellular compartments (nucleus, mitochondrial matrix, ER lumen, and ER membrane) revealed variations in absolute and relative activity [3]. TurboID signal was clearly detectable after 10 minutes in each compartment and was even stronger than BioID's 18-hour labeling in the mitochondrial matrix and ER lumen [3].
Model System Compatibility
The suitability of a PL enzyme depends heavily on the experimental model system. Key compatibility factors include toxicity, substrate permeability, and required labeling time.
Table 2: Enzyme Compatibility Across Model Systems
| Model System | Recommended Enzymes | Experimental Support & Key Considerations |
|---|---|---|
| Mammalian Cell Cultures | APEX2, TurboID, miniTurbo, BioID | APEX2 works well in cultured cells [32]. TurboID enables 10-min labeling [3]. H₂O₂ toxicity for APEX2 is manageable in cell culture. |
| Plants | TurboID, miniTurbo, BioID2 | TurboID enables proteomic mapping with low-abundant baits and in different plant species [28] [32]. APEX is less common due to background from endogenous peroxidases and biotin-phenol permeability issues [32]. |
| Microorganisms | TurboID, BioID | The high activity of TurboID makes it suitable for organisms where traditional BioID showed low efficiency [31] [3]. |
| Live Animals (e.g., Flies, Worms) | TurboID | TurboID extends biotin-based PL to flies and worms, systems where BioID was ineffective due to slow kinetics [3]. |
Detailed Experimental Workflows
A successful PL experiment requires careful design and execution. The following workflow, derived from established protocols, outlines the key steps for a TurboID experiment, which is widely applicable across model systems.
Diagram 1: PL Experimental Workflow
Protocol for TurboID-based Proximity Labeling
Step 1: Construct Design and Validation
- Fuse the TurboID enzyme to your protein of interest (bait) at either the N- or C-terminus [32].
- Critical Control: Express the TurboID enzyme alone or fused to an irrelevant localization peptide under the same promoter as the bait construct. This controls for background labeling and identifies proteins that are non-specifically labeled or bind to the beads [32].
- Functional Validation: Confirm that the bait-PL fusion protein is functional and localizes correctly within the cell. Functional complementation analysis is a prerequisite to ensure the fusion does not disrupt the bait's native function [32].
Step 2: Expression in the Model System
- Express the construct in your model system (e.g., transfection for mammalian cells, generation of stable transgenic lines for plants or organisms) [32] [3].
- For quantitative MS, use isotopically distinct Tandem Mass Tags (TMT) to label peptides from different samples, allowing for relative quantification across conditions [3].
Step 3: Biotin Application and Labeling
- Add biotin to the system to initiate labeling. For TurboID in cell culture, a concentration of 50-500 μM is typical [3].
- The labeling time can be as short as 10 minutes for TurboID, but this should be optimized for the specific bait and system [32] [3].
Step 4: Cell Lysis and Streptavidin Affinity Purification
- Lyse cells using a stringent RIPA buffer to ensure complete disruption.
- Incubate the lysate with streptavidin-coated beads to capture biotinylated proteins. The high affinity of the biotin-streptavidin interaction (Kd = 10⁻¹⁴ mol/L) allows for stringent washing under denaturing conditions (e.g., high detergent, salt, or denaturing agents) to reduce contaminants [32].
Step 5: On-bead Digestion and Mass Spectrometry Analysis
- Digest the captured proteins on the beads with trypsin.
- Analyze the resulting peptides via Liquid Chromatography-tandem Mass Spectrometry (LC-MS/MS) [32] [3].
- Data Analysis: Identify proteins significantly enriched in the bait sample compared to the control sample(s). ROC analysis using curated true-positive and false-positive protein lists for specific organelles can be used to filter results [3].
Advanced Applications and Emerging Methods
PL technology has expanded beyond basic proteomic mapping. Key innovations include:
- Split-Systems: Split-TurboID and Split-BioID allow conditional proteomics, where labeling only occurs when two bait proteins interact, bringing the split enzyme fragments together [21].
- RNA-Protein Interactions: Techniques like RaPID use a promiscuous biotin ligase (BASU) tethered to specific RNA motifs to identify binding proteins [29].
- Super-Resolution PL: New methods focus on identifying specific biotinylation sites (the modified lysine residues) rather than just enriched proteins. This provides direct evidence of labeling, reduces false positives, and can reveal membrane protein topologies [33]. One such method demonstrated an 88.2% true positive rate for mitochondrial matrix proteins without the need for a negative control, simplifying experimental design [33].
Essential Research Reagent Solutions
The table below lists key reagents required for a successful PL-MS experiment.
Table 3: Essential Research Reagents for Proximity Labeling
| Reagent / Tool | Function / Description | Example Use Case |
|---|---|---|
| TurboID Plasmid [3] | Genetically encoded biotin ligase for fast labeling. | The primary engine for proximity-dependent biotinylation in live cells. |
| Biotin [3] | Small molecule substrate for biotin ligases. | Added to the culture medium or organism to initiate the labeling reaction. |
| Streptavidin Beads [32] | High-affinity resin for purifying biotinylated proteins. | Used to capture and purify biotin-tagged proteins from a complex cell lysate. |
| Stringent Lysis/Wash Buffers [32] | Typically RIPA buffer; removes non-specifically bound proteins. | Ensures clean samples for MS by reducing background contaminants. |
| TMT (Tandem Mass Tags) [3] | Isobaric labels for multiplexed quantitative proteomics. | Allows simultaneous quantification of proteins from multiple conditions in a single MS run. |
| LC-MS/MS System | Instrumentation for peptide separation, fragmentation, and identification. | The core platform for identifying and quantifying the enriched proteome. |
Choosing the optimal proximity labeling enzyme requires balancing labeling speed, toxicity, and model system compatibility. TurboID and miniTurbo represent significant advances for applications in plants, microorganisms, and live animals where speed and minimal toxicity are critical. For controlled mammalian cell culture systems where ultimate temporal resolution is needed, APEX2 remains a powerful option. Future developments, particularly in site-specific identification and split-enzyme systems, promise to further enhance the precision and scope of proximity labeling, enabling researchers to dissect complex molecular networks with ever-greater accuracy across diverse biological contexts.
Proximity labeling (PL) has emerged as a revolutionary technology for mapping the spatial organization of proteomes within living cells. This technique enables researchers to capture intricate molecular relationships in challenging subcellular environments such as mitochondria, synapses, and organelle contact sites—compartments that have traditionally been difficult to study with conventional methods like affinity purification or yeast two-hybrid systems [8]. By generating reactive biotin species that covalently tag nearby proteins, PL provides a snapshot of the molecular neighborhood within a defined radius of a bait protein, allowing for the identification of transient interactions, membrane protein topologies, and localized proteomes with unprecedented spatial resolution [28] [19]. This comparison guide examines the performance of major PL enzymes and their applications across different biological contexts, providing researchers with evidence-based recommendations for selecting optimal tools for their experimental needs.
Comparing Proximity Labeling Enzymes
The effectiveness of proximity labeling experiments depends significantly on selecting the appropriate enzyme for the biological question and experimental system. The table below compares the key operational characteristics of major PL enzymes:
Table 1: Comparison of Major Proximity Labeling Enzymes
| Enzyme | Type | Labeling Time | Labeling Radius | Key Advantages | Major Limitations |
|---|---|---|---|---|---|
| BioID | Biotin Ligase | 12-16 hours [34] | ~10 nm [28] | Low background; suitable for in vivo applications [34] | Long labeling time misses transient interactions [28] |
| BioID2 | Biotin Ligase | Several hours [2] | ~10 nm [28] | Smaller size minimizes steric interference [2] | Still requires hours for labeling [2] |
| TurboID | Biotin Ligase | 10 minutes - 1 hour [28] [34] | ≥35 nm (time-dependent) [28] | Extremely fast labeling; broad temperature range [28] | High background; cellular toxicity [28] [2] |
| miniTurbo | Biotin Ligase | 10 minutes - 1 hour [28] | Similar to TurboID [28] | Fast labeling with lower background than TurboID [28] | Half as active as TurboID [28] |
| APEX/APEX2 | Peroxidase | 1 minute [28] | <20 nm [28] | Extremely fast; excellent temporal control [28] | Requires toxic H₂O₂; limited membrane permeability [28] |
| HRP | Peroxidase | Minutes [34] | 200-300 nm [34] | Extensive labeling radius [34] | Limited to extracellular/oxidizing environments [28] |
Recent engineering efforts have produced specialized PL enzymes with enhanced capabilities. AirID, a synthetic biotin ligase developed through ancestral enzyme reconstruction, shows greatly enhanced activity (1-6 hour labeling time) and broader temperature range [28]. microID and ultraID represent smaller BirA* variants with kinetics similar to TurboID but reduced background labeling from endogenous biotin [28]. For neuroscience applications, LOV-TurboID incorporates a light-sensitive LOV domain that enables spatiotemporal control via low-intensity blue light activation, significantly reducing background labeling in biotin-rich environments such as neurons [19].
Application-Specific Enzyme Performance
Mitochondrial Proteome Mapping
Mitochondrial studies have particularly benefited from PL technologies, especially for characterizing poorly understood processes like mitochondrial co-translational import [35]. In this process, translation is coupled to mitochondrial protein translocation, reducing the energy cost associated with post-translational import relying on chaperone systems [35]. When studying the TOM20 proxisome using BioID, researchers observed high enrichment of RNA binding proteins near the TOM complex, suggesting their potential involvement in co-translational import mechanisms [35].
APEX2 has proven exceptionally valuable for mitochondrial research due to its small labeling radius (<20 nm) and rapid labeling time (1 minute), which provides near-snapshot capability of protein interactions [35]. This has enabled precise mapping of mitochondrial matrix proteomes and resolution of membrane protein topology [35]. A comparative study evaluating APEX2 for mitochondrial matrix proteome mapping demonstrated that biotinylation site identification methods identified 449 true positive mitochondrial proteins with an 88.2% true positive rate, significantly outperforming conventional approaches [33].
Table 2: Proximity Labeling Applications in Mitochondrial Research
| Application | Recommended Enzyme | Key Findings | Experimental Considerations |
|---|---|---|---|
| Matrix Proteome | APEX2 [35] | Identified ~500 matrix proteins with high specificity [33] | Use in closed compartments; minimal radical permeability [35] |
| Surface Interactions | BioID [35] | Enriched RNA-binding proteins near TOM complex [35] | Longer labeling captures weaker associations [35] |
| Membrane Topology | APEX2 [35] | Resolved protein orientation in membranes [35] | Targeted to specific membrane faces [35] |
| Contact Sites | Split-TurboID [2] | Identified MAM proteome [35] | Requires reconstitution of enzyme fragments [2] |
Synaptic Proteome Analysis
The synaptic cleft represents a particularly challenging environment for proteomic mapping due to its extracellular location, protein density, and transient interactions [34] [36]. Traditional methods like biochemical fractionation struggle to preserve the native organization of synaptic proteins and often miss transient interactions [2]. PL technologies have enabled unprecedented insights into the molecular architecture of synapses by capturing protein interactions in living neurons under physiological conditions [34] [2].
BioID and its derivatives have been successfully applied to map the proteome of specific synapse types, synaptic clefts, and glial-neuronal interfaces [34]. In one pioneering study, PL facilitated the discovery of proteomes associated with specific neuronal populations, synaptic clefts, and tripartite synapses formed by astrocyte-neuron connections [34]. TurboID has proven valuable for capturing activity-dependent changes in synaptic protein composition due to its rapid labeling capability, though researchers must carefully optimize labeling conditions to minimize background in the biotin-rich neuronal environment [2].
For extracellular synaptic proteins, HRP-based labeling offers advantages due to its large labeling radius (200-300 nm) and compatibility with the oxidizing environment of the synaptic cleft [34]. However, HRP is ineffective for intracellular synaptic components due to its requirement for disulfide bonds that cannot form in the reducing intracellular environment [28] [34].
Organelle Contact Sites
Membrane contact sites between organelles represent critical hubs for cellular signaling, lipid transfer, and metabolic coordination, but their dynamic nature has made them difficult to study with traditional methods [37]. Recent advancements in PL have enabled systematic mapping of these transient interfaces, revealing previously unknown protein components and organizational principles.
The BiFCPL (Bimolecular Fluorescence Complementation-based Proximity Labeling) system represents a particularly innovative approach for studying organelle contact sites [37]. This strategy uses bimolecular fluorescence complementation to ensure that labeling only occurs when two organelles are in close proximity, enabling specific analysis of contact site proteomes in living cells [37]. When applied to mitochondria-endoplasmic reticulum contacts (MERCs), BiFCPL identified 403 high-confidence MERC proteins, including transiently resident proteins and potential tethers [37].
Similarly, application of BiFCPL to mitochondria-lipid droplet (LD) contacts revealed that these interfaces are highly sensitive to nutrient status [37]. A comparative proteomic analysis identified 60 proteins that are up- or downregulated at these contact sites under metabolic challenge, including SQLE, an enzyme for cholesterol synthesis that accumulates at mitochondria-LD contact sites probably to utilize local ATP for cholesterol synthesis [37].
Split-enzyme systems like Split-TurboID have also proven valuable for studying organelle contacts [2]. In these systems, the TurboID enzyme is split into two fragments that are targeted to different organelles or proteins; functional enzyme reconstitution only occurs when these compartments are in close proximity, enabling highly specific labeling of contact site proteomes [2].
Advanced Methodological Considerations
Experimental Workflow for Proximity Labeling
The diagram below illustrates the general workflow for a proximity labeling experiment, from enzyme selection to data analysis:
Detection Methodologies: Protein-Level vs. Peptide-Level Enrichment
A critical advancement in PL methodology has been the shift from protein-level to peptide-level enrichment approaches. Conventional protein-level enrichment methods often co-purify unlabeled peptides or indirectly associated proteins, leading to potential false positives [2]. Peptide-level enrichment enables direct identification of biotinylation sites, providing strong evidence that proteins were truly labeled in situ [2]. This site-specific information increases confidence in identifying true interactors and eliminates the need for negative control-based fold-change calculations [2].
A recently developed super-resolution proximity labeling method that directly identifies biotinylation sites demonstrated significant advantages over conventional ratiometric approaches [33]. When applied to mitochondrial matrix proteome mapping, this method identified 449 true positive mitochondrial proteins with an 88.2% true positive rate, compared to 471 true positives (78.8% true positive rate) using conventional methods [33]. The biotin-site identification method also showed 89% quantitative composition of true positives within the dataset, compared to only 24-36% for conventional approaches [33].
Research Reagent Solutions
Table 3: Essential Research Reagents for Proximity Labeling Experiments
| Reagent Category | Specific Examples | Function and Application |
|---|---|---|
| PL Enzymes | BioID, TurboID, APEX2 [28] [8] | Catalyze proximity-dependent biotinylation of nearby proteins |
| Biotin Substrates | Biotin, Biotin-phenol [28] | Enzyme substrates that generate reactive biotin species |
| Enrichment Matrices | Streptavidin beads, NeutrAvidin, Tamavidin 2-REV [34] [33] | Affinity capture of biotinylated proteins/peptides |
| Detection Reagents | Fluorescent streptavidin, Anti-biotin antibodies [34] [4] | Visualization and detection of biotinylation patterns |
| Activation Reagents | Hydrogen peroxide (for peroxidases) [28], Blue light (for LOV-TurboID) [19] | Enzyme activation for controlled labeling initiation |
Proximity labeling technologies have fundamentally transformed our ability to map subcellular proteomes in living cells, providing unprecedented insights into the spatial organization of mitochondrial, synaptic, and organelle contact site proteomes. The continuing evolution of PL enzymes—with improvements in labeling speed, specificity, and spatiotemporal control—promises to further enhance our understanding of dynamic cellular processes. As these tools become increasingly sophisticated, they will undoubtedly uncover new biological insights and accelerate drug discovery efforts targeting subcellular compartments in various disease contexts.
The optimal choice of PL enzyme remains highly context-dependent, requiring researchers to carefully consider their specific experimental needs regarding temporal resolution, spatial precision, and biological system compatibility. By matching the technical capabilities of each enzyme to the biological question, researchers can maximize the insights gained from their proximity labeling experiments.
Cellular signaling networks, particularly those mediated by G protein-coupled receptors (GPCRs) and kinases, rely on rapid, transient protein-protein interactions that have traditionally eluded conventional detection methods. Affinity purification mass spectrometry (AP-MS), the historical standard for identifying protein-protein interactions (PPIs), suffers from critical limitations when applied to these dynamic systems. The milder lysis conditions required often impede the capture of membrane proteins, and weaker or more transient interactions are frequently lost during the extraction process [18]. Given that synapses, GPCR signaling complexes, and kinase-substrate relationships are characterized by highly transient and complex protein interactions, these limitations mean AP-MS cannot provide the high-resolution data needed to fully characterize these molecular landscapes [18].
Proximity labeling (PL) technologies have emerged as powerful alternatives that overcome these barriers by enabling the mapping of molecular interactions within living cells under near-physiological conditions. These techniques utilize engineered enzymes that generate reactive molecules to tag nearby proteins covalently, preserving interactions that would be disrupted by cell lysis [18]. This review provides a comprehensive comparison of PL enzymes and their applications in studying GPCR signaling and kinase pathways, offering researchers a framework for selecting optimal tools for capturing transient molecular events.
Proximity Labeling Enzymes: A Technical Comparison
PL techniques employ engineered enzymes fused to a protein of interest (POI) that catalyze the covalent tagging of nearby endogenous proteins with a biotin substrate. The biotinylated proteins can then be selectively enriched using streptavidin-coated beads and identified via mass spectrometry, enabling detailed mapping of protein interaction networks within their native cellular environment [18]. The major enzyme systems differ significantly in their mechanisms, kinetics, and optimal applications.
Table 1: Comparison of Major Proximity Labeling Enzymes
| Enzyme | Mechanism | Labeling Radius | Labeling Time | Key Advantages | Major Limitations |
|---|---|---|---|---|---|
| BioID [18] | Mutated E. coli biotin ligase (BirA*) leaks biotin-AMP | ~10 nm | 18-24 hours | Minimal background; works in many compartments | Long labeling time misses rapid dynamics |
| BioID2 [18] | Optimized biotin ligase from A. aeolicus | ~10 nm | Several hours | Smaller size; reduced biotin requirement | Still requires hours for labeling |
| APEX/APEX2 [18] [38] | Engineered peroxidase oxidizes biotin-phenol | <20 nm | 1 minute | Excellent temporal resolution; works in multiple compartments | H2O2 causes oxidative stress |
| TurboID [18] | Evolved E. coli biotin ligase with enhanced activity | ~10 nm (increases with time) | 10 minutes | Rapid labeling; high sensitivity | High background; cellular toxicity |
| miniTurbo [18] | Truncated version of TurboID | ~10 nm | 10 minutes | Rapid labeling with less background than TurboID | Lower activity than TurboID |
| Split-TurboID [18] [6] | TurboID split into complementary fragments | Dependent on reconstitution | Minutes after reconstitution | High specificity for genuine interactions | Requires protein-fragment reconstitution |
The critical distinction between peroxidase-based (APEX/APEX2) and biotin ligase-based (BioID, TurboID) systems lies in their activation mechanisms and temporal resolution. APEX/APEX2 generates extremely short-lived phenoxyl radicals (<1 ms) upon hydrogen peroxide addition, enabling precise minute-scale temporal control ideal for capturing rapid signaling events [38] [28]. In contrast, biotin ligase-based systems utilize biotin-AMP intermediates that diffuse to label proximal proteins, typically requiring longer labeling times though next-generation variants like TurboID have significantly improved kinetics [18] [28].
Application 1: Mapping Dynamic GPCR Signaling Networks
GPCRs constitute the largest family of membrane receptors in humans, regulating virtually all physiological processes and representing prime drug targets. However, their signaling complexity, including rapid activation dynamics, compartment-specific signaling, and biased agonism, has made them challenging to study with traditional methods [38].
Experimental Approach: APEX2-GPCR Fusion Strategy
The foundational experiment for GPCR PL involves fusing APEX2 to the receptor's C-terminus, typically separated by a flexible linker to minimize functional perturbation [38]. Cells stably expressing the fusion construct are pre-incubated with biotin-phenol for approximately one hour, followed by ligand stimulation for varying durations. The labeling reaction is initiated by adding hydrogen peroxide for exactly one minute, after which the reaction is rapidly quenched with a solution containing ascorbate, trolox, and sodium azide [38]. Biotinylated proteins are then enriched under denaturing conditions using streptavidin beads, digested into peptides, and analyzed via quantitative mass spectrometry, often using isobaric tagging (TMT) for precise multiplexed quantification across multiple time points and conditions [38].
Key Findings in GPCR Signaling
Application of PL to the angiotensin II type 1 receptor (AT1R) revealed remarkable insights into GPCR signaling dynamics. In unstimulated cells, AT1R-APEX2 preferentially labeled heterotrimeric G proteins, with the signal transducer Gαq showing the strongest enrichment among G proteins, confirming pre-coupled receptor-G protein complexes despite the classical paradigm of agonist-induced coupling [38]. Within minutes of angiotensin II stimulation, the labeling profile shifted dramatically to include endocytic machinery components including β-arrestin 2, AP-2, clathrin, FCHo protein, and intersectins, capturing the rapid transition from G protein signaling to receptor internalization [38].
Similar approaches applied to Gi-coupled receptors identified a network of Gαi1-specific signaling partners, including the unexpected identification of PDZ-RhoGEF (PRG) as a direct effector of active Gαi1 but not the highly homologous Gαi2, demonstrating the precise selectivity achievable with PL [39]. More recent applications to the luteinizing hormone receptor (LHR) have enabled minute-scale resolution of the receptor's interactome as it traffics through very early endosomes, identifying novel regulators like RAP2B and RAB38 that modulate receptor signaling and post-endocytic sorting [40].
Table 2: Key GPCR Proximity Labeling Studies and Findings
| GPCR Target | PL Enzyme | Key Biological Findings | Temporal Resolution |
|---|---|---|---|
| AT1R [38] | APEX2 | Pre-coupled G proteins; rapid recruitment of endocytic machinery | 1-20 minutes |
| Gi-coupled Receptors [39] | BioID2 | Identification of PDZ-RhoGEF as Gαi1-specific effector | 24 hours (constitutive active mutant) |
| LHR [40] | APEX2 | Minute-scale trafficking interactome; identification of RAP2B and RAB38 as regulators | 2-30 minutes |
| β2 Adrenergic Receptor [38] | APEX2 | Generalizability of platform across GPCR families | Multiple time points |
Application 2: Identifying Kinase-Substrate Relationships
Kinase-mediated phosphorylation represents one of the most widespread regulatory mechanisms in cell signaling, yet establishing direct kinase-substrate relationships remains challenging due to the transient nature of these interactions and the extensive crosstalk within kinase networks.
Experimental Approach: Phospho-APEX (pAPEX) Methodology
The Phospho-APEX (pAPEX) strategy combines APEX2 proximity labeling with phosphopeptide enrichment to identify phosphorylated proteins proximal to a kinase of interest [41]. Researchers create stable cell lines expressing APEX2-tagged kinases (e.g., MAPK1, PKA) and perform proximity labeling with biotinyl tyramide (500 μM) for one hour, with kinase activation (e.g., using EGF or forskolin) during the final five minutes [41]. Following one minute of H2O2 treatment and quenching, biotinylated proteins are enriched using streptavidin beads, digested, and then subjected to phosphopeptide enrichment using metal oxide chromatography before TMT-based quantitative mass spectrometry analysis [41]. This dual enrichment strategy specifically captures both proximity interactions and phosphorylation events, enabling the identification of candidate direct substrates.
Key Findings in Kinase Signaling
Application of pAPEX to MAPK1 and PKA in HEK293T and HCT116 cells successfully identified numerous known and novel substrates, including the validation of C15orf39 as a novel MAPK1 substrate [41]. Complementary approaches using BioID rather than APEX have also proven successful for kinases like casein kinase 2 (CK2) and PKA, with one study identifying 24 and 35 putative substrates for CK2 and PKA, respectively, by combining BioID with kinase perturbation and phosphorylation motif matching [42]. These studies demonstrate how PL techniques can overcome the challenge of indirect phosphorylation events in traditional phosphoproteomics by adding spatial constraint - only proteins that come in direct proximity to the kinase are considered candidate direct substrates.
Comparative Experimental Performance Data
The selection of an appropriate PL enzyme depends critically on the biological question, required temporal resolution, and cellular context. The following comparative data illustrates the practical performance differences between these systems in relevant experimental contexts.
Table 3: Quantitative Performance Comparison of PL Systems in Key Studies
| Application Context | Enzyme Used | Proteins Identified | Labeling Duration | Temporal Specificity |
|---|---|---|---|---|
| GPCR Signaling (AT1R) [38] | APEX2 | 1,242 proteins | 1 minute | Excellent (minute-scale resolution) |
| Gαi1 Interactome [39] | BioID2 | Not specified (multiple classes) | 24 hours | Moderate (constitutive activation) |
| Kinase Substrate ID (MAPK1) [41] | APEX2 | Not specified (known + novel substrates) | 1 hour (5 min activation) | Good (activation-specific) |
| Calcium-Activated Labeling [6] | Split-TurboID | Not specified (proteome-wide) | 10 minutes | Excellent (coincidence detection) |
| Neural Proteome Mapping [18] | TurboID | >1,000 synaptic proteins | 10 minutes - 1 hour | Good (minute-scale resolution) |
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of PL experiments requires specific reagents and optimization approaches. The following table outlines critical components for establishing these methodologies.
Table 4: Essential Research Reagents for Proximity Labeling Experiments
| Reagent / Resource | Function/Purpose | Example Specifications |
|---|---|---|
| APEX2-Tagged Kinase/GPCR Constructs [41] [38] | Bait protein for proximity labeling | Lentiviral vectors with CMV promoter, C-terminal APEX2 fusion |
| Biotin-Phenol (Biotinyl Tyramide) [38] [41] | Enzyme substrate for labeling | 500 μM working concentration in culture medium |
| Hydrogen Peroxide (H₂O₂) [38] [41] | Activator for peroxidase enzymes | 1 mM final concentration, 1 minute incubation |
| Quenching Solution [38] [41] | Stops labeling reaction | PBS with 10 mM sodium ascorbate, 5 mM trolox, 10 mM sodium azide |
| Streptavidin Magnetic Beads [41] | Enrichment of biotinylated proteins | 75 μL bead suspension per sample, 3-hour incubation |
| Isobaric Tags (TMT) [38] [41] | Multiplexed quantitative proteomics | 10- or 11-plex TMT reagents, 1-hour labeling |
| Triple-Stage Mass Spectrometry [38] | Accurate peptide quantification | TMT-SPS-MS3 to reduce ratio distortion |
Proximity labeling technologies have fundamentally transformed our ability to capture transient interactions in GPCR signaling and kinase pathways, moving beyond the limitations of traditional biochemical methods. The comparative data presented herein demonstrates that enzyme selection represents a critical decision point - APEX2 provides unparalleled temporal resolution for rapid signaling events, while BioID variants and TurboID offer advantages in sensitivity and compatibility with diverse biological systems.
Future methodological developments will likely focus on expanding the toolbox of conditionally activated PL systems, similar to the calcium-activated split-TurboID (CaST) approach [6], which could enable precise recording of cellular activity history in response to specific stimuli. Additionally, continued refinement of mass spectrometry methods, including peptide-level enrichment to directly identify biotinylation sites, will enhance specificity and reduce false positives in interaction datasets [18]. As these technologies mature and become more accessible, they will undoubtedly uncover new layers of complexity in cellular signaling networks and provide unprecedented insights into the dynamic molecular interactions that underlie physiology and disease.
Proximity labeling (PL) has revolutionized the study of biomolecular interactions by enabling the covalent tagging of proteins and other molecules in living systems. A particularly powerful evolution of this technology is the development of split-enzyme systems, which introduce an essential requirement for molecular reconstitution to trigger labeling. This configuration dramatically increases specificity by ensuring that labeling only occurs when two target proteins of interest come into close proximity, effectively recording transient interaction events that are difficult to capture with traditional methods.
This guide provides a comprehensive comparison of split-enzyme systems, focusing on their engineering principles, performance characteristics, and experimental applications. We objectively evaluate the leading platforms—split-TurboID and split-HaloTag—including quantitative data on their labeling kinetics, affinity ranges, and operational parameters to inform selection for specific research needs in intracellular interaction mapping.
Technology Comparison: Operational Principles and Performance Metrics
Split-enzyme systems function through a modular design where inactive fragments of a labeling enzyme are fused to different proteins of interest. Upon interaction between these target proteins, the enzyme fragments reconstitute into an active form, enabling localized labeling of nearby biomolecules. The table below compares the key characteristics of major split-enzyme systems.
Table 1: Comparison of Major Split-Enzyme Systems for Proximity Labeling
| System | Parent Enzyme | Activation Mechanism | Labeling Radius | Primary Substrate | Optimal Labeling Time | Key Advantages |
|---|---|---|---|---|---|---|
| Split-TurboID [6] [18] | TurboID (biotin ligase) | Ca²⁺-induced calmodulin/M13 peptide interaction [6] | ≥35 nm (time-dependent) [28] | Biotin | 10 minutes [6] | Rapid labeling; high sensitivity; works in deep tissues without light |
| CaST (Ca²⁺-activated split-TurboID) [6] | Split-TurboID | Ca²⁺-induced calmodulin/M13 peptide interaction | Not specified | Biotin | 10 minutes | Functions as coincidence detector for Ca²⁺ and biotin; reversible |
| Split-HaloTag [43] | HaloTag (hydrolase) | Peptide fragment (Hpep) binding to circularly permuted core (cpHalo∆) | Not applicable (direct covalent binding) | Chloroalkane (CA) ligands | 20 minutes to hours (improved with engineering) [43] | Permanent covalent tagging; multi-color imaging capabilities |
| Improved split-HaloTag (cpHalo∆2) [43] | Engineered HaloTag | Enhanced Hpep binding to stabilized cpHalo∆ | Not applicable (direct covalent binding) | Chloroalkane (CA) ligands | Significantly reduced vs. original [43] | 475x faster labeling; 15°C higher thermal stability |
Performance varies significantly across these systems, particularly regarding speed and temporal resolution. The following table summarizes critical quantitative performance data to guide experimental design.
Table 2: Quantitative Performance Metrics of Split-Enzyme Systems
| System | Apparent Second-Order Rate Constant (M⁻¹s⁻¹) | Affinity Range (Hpep EC₅₀) | Thermal Stability (Tm) | Calcium Sensitivity | Dynamic Range (Fold-Change) |
|---|---|---|---|---|---|
| Split-TurboID [6] | Not specified | Not applicable | Not specified | Yes (CaST variant) | 5.0 (CaST-IRES, +Ca²⁺ vs. -Ca²⁺) [6] |
| Original split-HaloTag [43] | ~1.0 × 10⁴ | Nano- to milli-molar | 31.8°C | Engineerable (via sensing domains) | Not specified |
| Improved split-HaloTag (cpHalo∆2) [43] | 1.61 × 10⁶ | 3.5x lower EC₅₀ vs. original [43] | 45.2°C | Engineerable (via sensing domains) | Not specified |
Experimental Protocols for Key Split-Enzyme Applications
CaST for Recording Calcium Signaling and Neuronal Activity
Background: Ca²⁺-activated split-TurboID (CaST) was engineered to biochemically record histories of cellular activation, particularly neuronal firing marked by intracellular calcium flux, in freely behaving animals without requiring fiber implants for light delivery [6].
Detailed Protocol:
- Construct Design and Expression: Utilize a bicistronic vector (CaST-IRES) expressing two fragments: (1) CD4-sTb(C)-M13-GFP (membrane-tethered) and (2) CaM-V5-sTb(N) (cytosolic). The IRES sequence ensures coordinated expression of both fragments in a controlled 5:2 ratio, which is critical for optimal signal-to-background ratio [6].
- Cell Transfection and Validation: Transfect cells (e.g., HEK293T) with the CaST-IRES construct. Validate expression and localization of both fragments using immunohistochemistry and confocal microscopy against the GFP and V5 tags [6].
- Activity-Dependent Labeling:
- Deliver biotin (500 µM) to the cell culture medium or systemically to animals. Biotin crosses the blood-brain barrier, enabling applications in the brain [6].
- Elevated intracellular Ca²⁺ induces calmodulin (CaM) binding to the M13 peptide. This reconstitutes split-TurboID activity.
- The reconstituted enzyme uses ATP to convert biotin into reactive biotin-AMP, which covalently tags lysine residues on proximal proteins (within ~10 nm radius) [28].
- A labeling time of 10 minutes is sufficient for robust detection [6].
- Reversibility Test (For Temporal Gating): To confirm that labeling only occurs during the biotin window, treat cells with Ca²⁺ and ionophore for 30 minutes, wash thoroughly for 10 minutes to return Ca²⁺ to baseline, and then add biotin for 30 minutes. This control should show minimal biotinylation, demonstrating the system's reversibility and precise temporal gating [6].
- Detection and Analysis:
- Immediate Readout: Fix cells and detect biotinylated proteins using streptavidin conjugated to Alexa Fluor 647 (SA-647) for fluorescence imaging. Normalize the SA-647 signal to the GFP fluorescence (SA-647/GFP ratio) to account for variation in tool expression across cells [6].
- Proteomic Analysis: Lyse cells and enrich biotinylated proteins with streptavidin-coated magnetic beads. Identify labeled proteins via liquid chromatography-mass spectrometry (LC-MS/MS) [18] [33].
Improved Split-HaloTag for Recording Transient Signaling Events
Background: Split-HaloTag records transient physiological events, such as GPCR signaling or calcium fluctuations, by transforming transient molecular interactions into permanent fluorescent marks. Recent engineering has addressed its initial limitation of slow labeling kinetics [43].
Detailed Protocol:
- System Components:
- cpHalo∆2: The large, engineered fragment with improved stability and activity.
- Hpep (1-8): A series of small peptide fragments with affinities for cpHalo∆2 ranging from nano- to milli-molar, allowing tuning based on application [43].
- Fluorescent Ligands: Cell-permeable chloroalkane (CA) substrates conjugated to fluorophores (e.g., TMR, JF dyes).
- Sensor Assembly for Calcium Recording:
- Fuse cpHalo∆2 to calmodulin (CaM) and the Hpep to the M13 peptide.
- During elevated calcium levels, CaM binds to M13, bringing Hpep into proximity with cpHalo∆2 and reconstituting the active HaloTag.
- Labeling and Detection:
- Add the CA-fluorophore ligand to the culture medium or administer it to live animals.
- The reconstituted HaloTag rapidly forms a stable covalent bond with the ligand, permanently tagging cells that were active during the ligand exposure window.
- With the improved cpHalo∆2, labeling at 37°C is significantly faster, requiring lower substrate concentrations and shorter incubation times (reduced from hours to minutes) for robust signal [43].
- Validation and Multi-Epoch Recording:
- Validate recording specificity using controls lacking the Hpep fragment or stimulus.
- For multi-epoch recording, sequentially administer CA ligands conjugated to different fluorophores (e.g., CA-JF549 and CA-JF646) to track distinct activity windows in the same cell population [43].
Signaling Pathways and Experimental Workflows
The following diagrams illustrate the core operational principles of two primary split-enzyme systems.
CaST Mechanism for Recording Calcium Activity
Split-HaloTag Workflow for Interaction Mapping
Research Reagent Solutions
The table below lists essential reagents for implementing split-enzyme proximity labeling experiments.
Table 3: Essential Research Reagents for Split-Enzyme Proximity Labeling
| Reagent / Material | Function / Role | Example Application / Note |
|---|---|---|
| Biotin | Small molecule substrate for biotin ligase-based systems (TurboID, split-TurboID). | Added to cell medium or administered in vivo; cell- and blood-brain barrier-permeable [6]. |
| Chloroalkane (CA) Ligands | Covalent substrate for HaloTag-based systems. | Conjugated to fluorophores for imaging or to handles for biochem. enrichment; enables multi-color pulse-chase [43]. |
| Biotin-Phenol (BP) | Substrate for peroxidase-based systems (APEX/APEX2). | Used with H₂O₂ for rapid labeling; less suitable for deep tissues due to H₂O₂ toxicity [19] [44]. |
| Streptavidin-Conjugated Beads | Enrichment of biotinylated proteins or peptides. | Critical for downstream MS-based proteomic analysis; magnetic beads facilitate washing [6] [33]. |
| Fluorescent Streptavidin (e.g., SA-647) | Direct imaging of biotinylation signals. | Allows immediate readout of labeling post-fixation; used for validation and quantification [6]. |
| Hpep Variants (1-8) | Small peptide fragments for split-HaloTag reconstitution. | Library of peptides with tuned affinities (nM-mM) allows experimental optimization [43]. |
| Calcium Ionophore (e.g., Ionomycin) | Artificial elevation of intracellular Ca²⁺. | Used as a positive control stimulus in calcium-sensing systems like CaST [6]. |
The field of intracellular mapping is undergoing a transformative shift from single-technology approaches to integrated multi-modal frameworks. While proximity labeling (PL) techniques like APEX2 and TurboID have revolutionized our ability to capture protein interactomes in living cells, researchers are increasingly recognizing that neither proteomics nor any other single methodology can fully capture the complexity of cellular organization. The integration of proteomics with transcriptomics and high-resolution electron microscopy (EM) now provides complementary layers of information that create a more comprehensive picture of cellular architecture and function. This comparative guide examines how these technologies perform when integrated, providing experimental data and protocols to help researchers select the optimal approach for their intracellular tagging research.
Comparative Performance of Integrated Methodologies
Table 1: Performance Metrics of Integrated Omics and Imaging Approaches
| Technology | Spatial Resolution | Temporal Resolution | Molecular Coverage | Key Advantages | Primary Limitations |
|---|---|---|---|---|---|
| PL + Transcriptomics | 10-20 nm (PL) + subcellular fractionation | Minutes (TurboID) to hours (BioID) | Proteins + RNA transcripts | Captures dynamic RNA-protein relationships; reveals post-transcriptional regulation | Limited spatial precision for RNA localization; may miss transient interactions |
| PL + Proteomics | 10-20 nm | Minutes (APEX2) to hours (BioID) | Protein-protein interactions + full proteome | Identifies direct interaction partners within complexes; functional validation | Endogenous biotinylation background; potential cytotoxicity with H₂O₂ |
| PL + EM | ≤10 nm (EM) + 10-20 nm (PL) | Fixed time points only | Ultrastructure + protein localization | Direct correlation of structure and molecular organization; nanoscale precision | Requires specialized sample preparation; cannot capture dynamics |
| Full Multi-Omics + EM | ≤10 nm to subcellular | Minutes to fixed points | Proteins, RNA, metabolites, ultrastructure | Most comprehensive view; validates findings across modalities | Technically challenging; resource-intensive; complex data integration |
Proximity Labeling Technologies: Engine Performance Comparison
Table 2: Proximity Labeling Enzymes for Multi-Modal Integration
| Enzyme | Size (kDa) | Labeling Time | Spatial Resolution | Best Suited Integration | Key Considerations for Integration |
|---|---|---|---|---|---|
| APEX2 | 28 | 1 minute | 10-20 nm | EM, Transcriptomics, Proteomics | Oxidative stress potential; compatible with EM staining |
| TurboID | 35 | 10 minutes | <10 nm | Transcriptomics, Proteomics | High background; endogenous biotin interference |
| BioID2 | 25 | 18-24 hours | 10 nm | Proteomics, Transcriptomics | Slow kinetics limits dynamic processes |
| HyPro2 | ~28 (modified) | Minutes | Single RNA molecule resolution | RNA-protein interactomics | Optimized for fixed cells; RNA-centric applications |
Experimental Protocols for Integrated Workflows
Protocol 1: Integrated Proximity Labeling and Transcriptomics
Workflow: MERR APEX-seq for Centrosomal Transcriptome Mapping [45]
Cell Line Engineering: Generate HEK293T cell line stably expressing APEX2-PCNT-EGFP fusion protein using lentiviral infection for centrosomal targeting.
Metabolic Pre-treatment: Incubate cells with 100 μM s6G (electron-rich nucleoside) for 5 hours to enhance RNA detection sensitivity.
Proximity Labeling:
- Add 0.5 mM biotin-phenol (BP) to culture medium for 30 minutes
- Initiate labeling with 1 mM H₂O₂ for exactly 1 minute
- Quench with scavenger cocktail (sodium ascorbate, sodium azide, and Trolox)
RNA Extraction and Purification:
- Lyse cells and extract total RNA
- Digest residual DNA with DNase I
- Isolate biotinylated RNAs using streptavidin-coated magnetic beads
- Select poly(A)+ RNA and fragment for cDNA library construction
Sequencing and Analysis: Perform high-throughput sequencing with DESeq2 analysis comparing experimental samples to cytosolic APEX2-NES controls.
Application: Protein interactome mapping of single RNA molecules
Cell Fixation and Permeabilization: Fix cells with 4% PFA for 15 minutes, then permeabilize with 0.1% Triton X-100.
Hybridization: Incubate with digoxigenin (DIG)-modified antisense oligonucleotides targeting specific RNA molecules.
Enzyme Recruitment: Add engineered HyPro2 enzyme containing DIG-binding domain and enhanced APEX2 derivative.
Proximity Biotinylation:
- Use optimized labeling buffer with 50% trehalose to limit diffusion
- Add biotin-phenol and H₂O₂ to trigger labeling (1-5 minutes)
- Quench with Trolox and ascorbate
Proteomic Analysis:
- Lyse cells and capture biotinylated proteins with streptavidin beads
- On-bead digestion with trypsin
- LC-MS/MS analysis and database searching
Application: Mitochondrial dysfunction analysis in aging hearts
Tissue Preparation: Collect cardiac tissue from mice at multiple age points (12, 24, 30 months).
Parallel Processing:
- EM Sample Prep: Fix with glutaraldehyde/OsO₄, dehydrate, embed in resin, section for EM
- Spatial Transcriptomics: Cryosection for spatial RNA-seq using 10x Genomics Visium
- Proteomics: Tissue homogenization, protein extraction, tryptic digestion, LC-MS/MS
- Metabo-lipidomics: Metabolite extraction with methanol/water, LC-MS analysis
Correlative Analysis:
- Quantify mitochondrial structural parameters from EM (size, cristae integrity)
- Correlate with proteomic data on mitochondrial proteins
- Integrate with spatial transcriptomics for subregional expression patterns
- Validate with metabo-lipidomics for oxidative stress markers
Integrated Data Analysis: Resolving Molecular Relationships
The integration of transcriptomic and proteomic data reveals complex relationships between mRNA availability and protein abundance, highlighting the importance of multi-modal approaches. In hippocampal mapping, researchers found that while many transcripts and proteins show correlated spatial enrichment, significant decoupling occurs due to factors including protein half-life differences, local translation, and trafficking mechanisms [46].
Spatial transcript-protein relationships in neurons show complex regulatory mechanisms beyond linear mRNA-to-protein translation [46].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagent Solutions for Integrated Proximity Labeling Studies
| Reagent/Category | Specific Examples | Function in Workflow | Integration Considerations |
|---|---|---|---|
| Proximity Enzymes | APEX2, TurboID, HyPro2, BioID2 | Catalyze biotinylation of proximal biomolecules | Size, kinetics, and compatibility with fixation protocols vary |
| Biotin Substrates | Biotin-phenol, Biotin-AMP | Reactive donors for proximity labeling | Membrane permeability and radical diffusion range critical |
| Quenching Reagents | Trolox, Sodium ascorbate, Azide | Terminate labeling reaction; reduce background | Must preserve RNA/protein integrity for downstream assays |
| Viscosity Agents | Trehalose, Sucrose | Limit diffusion of reactive species | Trehalose (50%) optimizes labeling specificity without significant activity loss |
| Sensitivity Enhancers | s6G, s4U nucleosides | Metabolic RNA tagging for improved detection | Enable transcriptome mapping in small compartments |
| Validation Tools | EM, RNA-FISH, Immunofluorescence | Corroborate spatial localization and interactions | Provide orthogonal verification of omics findings |
Performance Optimization Strategies
Enhancing Spatial Specificity
The challenge of biotin diffusion in proximity labeling is particularly relevant when studying small compartments like transcription sites or individual RNA molecules. Research demonstrates that adding 50% trehalose to the labeling buffer significantly reduces diffusion artifacts while maintaining enzyme activity better than sucrose-based alternatives [4]. This optimization is crucial when integrating with EM or transcriptomics where precise spatial information is paramount.
Multi-Omic Data Integration Frameworks
Successful integration requires specialized bioinformatic approaches:
- Cross-referencing spatial enrichment patterns between transcriptomic and proteomic datasets
- Pathway enrichment convergence analysis to identify biologically relevant signals
- Protein-RNA correlation assessment to understand post-transcriptional regulation
- Structural correlation between EM features and molecular abundance patterns
The choice between integration strategies depends heavily on research priorities. For dynamic process analysis in living cells, PL with transcriptomics offers insights into rapid regulatory mechanisms. For structural studies, PL with EM provides nanoscale resolution of molecular positioning. The most comprehensive understanding emerges from full multi-modal integration, though this requires substantial resources and computational expertise. As these technologies continue to evolve, particularly with emerging enzyme-free and light-activated labeling systems, the resolution and scope of integrable intracellular mapping will further expand, offering unprecedented views of cellular organization across molecular scales.
Researchers should carefully consider their specific biological questions, required resolution, and available resources when selecting an integration strategy, recognizing that each approach offers complementary strengths for decoding the intricate organization of cellular systems.
Navigating Technical Challenges: A Guide to Robust and Reproducible PL Experiments
Proximity labeling (PL) has revolutionized the study of biomolecular interactions by enabling the covalent tagging and identification of proteins in the immediate environment of a target protein of interest (POI) in living cells [18]. However, the accuracy and interpretation of these experiments are highly dependent on the use of rigorous controls. Properly designed negative and spatial controls are not merely optional but are fundamental to distinguishing specific interactions from background noise and to drawing meaningful biological conclusions. This guide examines the critical controls required for intracellular tagging research, comparing the performance of different PL enzymes and providing supporting experimental data.
The Proximity Labeling Toolkit: Enzymes and Mechanisms
Proximity labeling enzymes can be broadly categorized into two families: peroxidases (e.g., APEX/APEX2) and biotin ligases (e.g., BioID, TurboID). Their distinct mechanisms of action directly influence experimental design, particularly the choice of controls.
The following diagram illustrates the core catalytic mechanisms of these two major enzyme classes:
Comparison of Proximity Labeling Mechanisms
Comparative Performance of PL Enzymes
The choice of enzyme is critical and involves trade-offs between labeling radius, temporal resolution, and practical considerations like toxicity. The table below summarizes the key characteristics of widely used PL enzymes:
| Enzyme | Catalytic Mechanism | Labeling Time | Labeling Radius (Est.) | Key Advantages | Key Limitations |
|---|---|---|---|---|---|
| APEX2 | Peroxidase; Uses H₂O₂ to oxidize biotin-phenol into phenoxyl radicals [18] | 1 minute to 30 minutes [18] [47] | 10-20 nm [18] | High temporal resolution; works in multiple cellular compartments [47] | Requires toxic H₂O₂; can cause oxidative stress [18] |
| TurboID | Mutant biotin ligase; utilizes ATP to generate reactive biotin-AMP [18] [47] | 10 minutes to 6-24 hours (highly context-dependent) [18] [6] [48] | < 10 nm to ~100 nm [18] | Extremely high catalytic activity; requires only biotin delivery [47] | High background labeling if not optimized; can affect cell viability [18] |
| BioID | Mutant biotin ligase; generates reactive biotin-AMP [18] | 18-24 hours [18] | ~10 nm [18] | Low background; well-established protocol | Very slow catalysis, poor for capturing dynamic processes [18] |
| HyPro2 | Peroxidase; enhanced version of APEX for RNA-protein interactomes [4] | Not Specified | Not Specified | Improved labeling efficiency for low-abundance RNA targets [4] | Specialized for RNA-centric applications; requires fixation [4] |
A Framework for Critical Controls in PL Experiments
A well-controlled PL experiment requires multiple layers of validation to ensure that the identified interactors are specific to the POI and its correct subcellular localization.
Negative Controls for Specificity
Negative controls are essential to identify proteins that are biotinylated non-specifically. The most common strategies are compared in the table below:
| Control Type | Description | Experimental Example | Strength of Evidence | Limitations |
|---|---|---|---|---|
| Catalytic Dead Enzyme | Expressing a catalytically inactive mutant of the PL enzyme fused to the POI [47] | Using APEX2 with a mutated active site [47] | High. Controls for background from enzyme expression and biotin handling. | Does not account for potential "neighborhood" effects caused by the physical presence of the POI. |
| No Substrate Control | Expressing the active PL enzyme but omitting a key substrate (e.g., H₂O₂ for APEX2 or biotin for TurboID) [47] | Omitting H₂O₂ or biotin-phenol in an APEX2 experiment [47] | High. Essential for identifying proteins that bind non-specifically to streptavidin beads. | Must be used in conjunction with other controls. |
| Cytosolic Enzyme | Expressing the active PL enzyme targeted to the cytosol (or another irrelevant compartment) [48] | Pan-neuronal expression of cytosolic TurboID in C. elegans [48] | Moderate. Useful for general background, especially in in vivo models. | Low spatial specificity; may be too permissive as it does not control for the POI's microenvironment [48]. |
| Spatial Mislocalization | Expressing the active PL enzyme fused to a POI that is mutated to mislocalize within the cell [48] | Deleting the C-terminal PDZ-binding motif (PBM) of neurexin, causing its dispersal from presynaptic active zones [48] | Very High. The most rigorous control; identifies interactions dependent on the POI's correct localization [48]. | Requires prior knowledge of the targeting domain of the POI. |
The following workflow integrates these control strategies into a robust experimental design for data interpretation:
Workflow for Controlled Proximity Labeling Experiment
Spatial Controls for Compartment-Specific Resolution
Spatial controls help refine the proteomic map to a specific organelle or subcellular compartment. Ratiometric tagging is a powerful quantitative spatial control strategy.
- Principle: This involves comparing labeling patterns when the same PL enzyme is targeted to two different, closely related compartments (e.g., the mitochondrial matrix vs. the cytosol) [47].
- Implementation: Using stable isotope labeling with amino acids in cell culture (SILAC) or tandem mass tag (TMT) labeling, proteins are quantified from both experiments. A high ratio of labeling in the target compartment (e.g., matrix) versus the reference compartment (e.g., cytosol) provides strong evidence for specific localization [47]. This method was successfully used to map the mitochondrial intermembrane space proteome with high specificity [47].
Case Studies in Control Design
Case Study 1: Mapping the Neurexin Interactome with a Localization Control
A study mapping the intracellular interactors of the synaptic adhesion protein neurexin in C. elegans provides a prime example of an optimized spatial control [48].
- Experimental Strain: Endogenously tagged neurexin with TurboID at its intracellular domain, which preserved its function and presynaptic localization [48].
- Critical Control Strain ("ΔPBM"): The C-terminal PDZ-binding motif (PBM) of neurexin was deleted in the TurboID-tagged strain. This caused neurexin to disperse along the cell surface instead of clustering at synapses, while the enzyme remained functional [48].
- Outcome: By comparing the experimental strain to the ΔPBM control, researchers identified proteins that specifically interacted with neurexin at the synapse. This control was deemed more specific than using a wild-type strain or a cytosolic TurboID expression control, which were either too restrictive or too permissive, respectively [48].
Case Study 2: Capturing Calcium-Dependent Interactions with an Orthogonal Trigger
The development of Ca²⁺-activated split-TurboID (CaST) demonstrates the use of a functional or orthogonal control [6].
- Mechanism: CaST is a split enzyme where the two halves reconstitute only in the presence of high intracellular Ca²⁺. Biotinylation, therefore, requires two coincident signals: elevated Ca²⁺ and the presence of exogenous biotin [6].
- Inherent Controls: The primary negative controls are:
- Cells with high Ca²⁺ but no exogenous biotin (no signal due to lack of substrate).
- Cells with exogenous biotin but resting Ca²⁺ levels (no signal because the enzyme is split and inactive) [6].
- Application: This system was used to tag neurons in the mouse prefrontal cortex activated by psilocybin, with high temporal resolution and minimal background [6].
Essential Research Reagent Solutions
The table below lists key reagents critical for successfully executing controlled PL experiments.
| Reagent / Tool | Function in Experiment | Key Consideration |
|---|---|---|
| TurboID / APEX2 Plasmids | Genetically encoded fusion tags for proximity labeling. | Select based on required speed, compartment compatibility, and toxicity profile [18] [47]. |
| Biotin (for TurboID/BioID) | Substrate for biotin ligase-based PL. | Concentration and incubation time must be optimized to balance signal and background [18] [48]. |
| Biotin-Phenol & H₂O₂ (for APEX2) | Substrates for peroxidase-based PL. | H₂O₂ is cytotoxic; concentration and pulse time must be carefully titrated [18] [47]. |
| Streptavidin-conjugated Beads | To affinity-purify biotinylated proteins after cell lysis. | High-binding capacity and purity are essential for efficient pull-down and clean MS data. |
| SILAC or TMT Kits | For quantitative mass spectrometry, enabling ratiometric comparison of different conditions (e.g., experimental vs. control) [47]. | Allows for precise quantification and statistical confidence in identifying specific interactors [47]. |
| Validated Negative Control Constructs | e.g., Catalytically dead enzyme, cytosolic-localized PL, or mislocalized POI constructs. | The choice of control should be tailored to the biological question and the POI's biology [47] [48]. |
The power of proximity labeling to map the cellular interactome is inextricably linked to the rigor of its experimental controls. No single control is perfect, and a combination of strategies—such as using a catalytic dead mutant alongside a spatial mislocalization control—often yields the most reliable and interpretable data. As the field evolves with new enzymes like HyPro2 [4] and condition-activated tools like CaST [6], the principles of careful control design remain the bedrock upon which meaningful biological discoveries are built.
Proximity labeling (PL) has revolutionized the study of protein-protein interactions and subcellular proteomes by enabling the covalent tagging of neighboring proteins in living cells [2]. However, the widespread adoption of PL techniques across diverse biological systems has revealed a critical and pervasive challenge: high background signals from endogenous biotinylation and nonspecific binding that can compromise data quality and interpretation [49] [33] [11]. This background noise presents a significant obstacle for researchers seeking to identify genuine interactors with high confidence, particularly when studying low-abundance proteins or complex molecular environments.
The fundamental issue stems from multiple sources. Endogenously biotinylated proteins, particularly mitochondrial carboxylases, consistently appear as high-abundance background in streptavidin-based purifications [49] [2]. Additionally, nonspecific binding of proteins to streptavidin beads, labeling outside the desired subcellular compartment, and diffusion of reactive intermediates all contribute to background signals that can obscure true biological interactions [50] [11]. The severity of these background issues varies considerably across different PL enzymes, biological systems, and experimental conditions, necessitating a sophisticated understanding of their underlying causes and appropriate mitigation strategies.
This guide provides a comprehensive comparison of background challenges across major PL platforms and presents empirically validated strategies to enhance signal-to-noise ratios. By systematically addressing the sources of background and implementing optimized experimental designs, researchers can significantly improve the specificity and reliability of their proximity labeling results across diverse applications.
The background signals in PL experiments arise from distinct mechanisms that vary in significance depending on the enzymatic platform and biological context. Endogenous biotinylation represents a universal challenge, as biotin-dependent carboxylases in mitochondria (e.g., propionyl-CoA carboxylase, methylcrotonyl-CoA carboxylase) are abundantly expressed and create strong signals that can dominate mass spectrometry analyses [49] [2]. These proteins are covalently modified with biotin in normal cellular metabolism and are co-purified with experimentally biotinylated proteins during streptavidin capture.
Nonspecific binding to streptavidin beads represents another major source of background, particularly for hydrophobic membrane proteins and highly abundant cellular proteins that may bind to the streptavidin matrix or plasticware independent of biotin modification [33]. The extreme affinity of the biotin-streptavidin interaction (Kd ~ 10-14 M) enables highly stringent washing conditions that reduce but do not eliminate this background [49].
Enzyme-specific artifacts constitute the third major category. For biotin ligases like TurboID, basal activity in the presence of endogenous biotin can cause labeling before experimental initiation [3] [2]. For peroxidase-based systems like APEX, endogenous cellular peroxidases can utilize hydrogen peroxide to catalyze background labeling, particularly in certain cell types such as C2C12 myoblasts, 3T3-L1 pre-adipocytes, and NIH/3T3 fibroblasts [11].
Platform-Specific Background Profiles
Table 1: Comparison of Background Sources Across Major Proximity Labeling Platforms
| Platform | Major Background Sources | Typical Labeling Time | Key Background Limitations |
|---|---|---|---|
| BioID | Endogenous biotinylated proteins, nonspecific bead binding | 18-24 hours | Low catalytic efficiency requires long labeling times, increasing stochastic background [3] |
| TurboID | Endogenous biotinylated proteins, basal enzyme activity, nonspecific binding | 10 minutes | High activity causes background with endogenous biotin; cellular toxicity concerns [3] [2] |
| APEX/APEX2 | Endogenous peroxidases, H2O2 toxicity, nonspecific binding | 1 minute | H2O2 addition activates endogenous peroxidases; toxic to sensitive cells and tissues [11] [2] |
| iAPEX | Reduced endogenous peroxidase background, minimal H2O2 toxicity | 1 minute (with DAAO activation) | Requires two-component system; limited to compartments with targeted DAAO [11] |
| LaccID | Nonspecific binding, media components | 1-2 hours | Lower catalytic efficiency than APEX/HRP; inhibited by culture media thiols [51] |
The table above illustrates how different PL systems exhibit distinct background profiles. Biotin ligase-based methods primarily struggle with interference from endogenous biotinylation, while peroxidase-based systems face challenges from endogenous peroxidase activity and H2O2 toxicity. A recent study found that in some cell types, background biotinylation from endogenous peroxidases can surpass the specific signal from APEX2, rendering conventional APEX labeling ineffective without additional countermeasures [11].
Systematic Strategies for Background Reduction
Experimental Design and Normalization Approaches
Strategic Control Design is foundational for distinguishing specific labeling from background. The most effective approach involves comparing samples expressing the bait-fused PL enzyme against multiple controls: (1) untransfected cells, (2) cells expressing the PL enzyme alone or mislocalized to a different compartment, and (3) samples where labeling is omitted by withholding biotin (for biotin ligases) or H2O2 (for peroxidases) [47] [2]. This multi-pronged control strategy enables statistical discrimination of true interactors from background proteins.
Quantitative Proteomic Normalization methods can significantly reduce technical variations. Research demonstrates that normalizing PL data to endogenously biotinylated proteins (such as PCCA) minimizes batch effects and enables fair comparisons across different experiments and PL probes [49]. For APEX-based experiments, ratiometric tagging strategies that compare labeling in multiple compartments (e.g., target compartment versus cytosol) have successfully mapped proteomes of challenging cellular regions like the mitochondrial intermembrane space [47].
Enzyme Cascade Systems represent an innovative approach to reduce background in peroxidase-based labeling. The recently developed iAPEX (in situ APEX activation) system combines APEX2 with D-amino acid oxidase (DAAO) to locally produce H2O2 only where needed, minimizing global activation of endogenous peroxidases [11]. This system reduces oxidative stress and expands APEX applications to cell types previously incompatible with conventional APEX labeling due to high background.
Technical Optimization at Critical Workflow Stages
Biotinylation Reaction Optimization requires careful titration of key parameters. For TurboID, optimizing biotin concentration (typically 50-500 μM) and labeling time (10 minutes to several hours) can balance labeling efficiency against background [3] [2]. For APEX2, minimizing H2O2 exposure time (1 minute or less) reduces both toxicity and nonspecific labeling [2]. For the emerging LaccID system, using appropriate media (Earle's Balanced Salt Solution instead of DMEM) and optimized substrates like biotin-methoxyphenol rather than standard biotin-phenol can enhance signal-to-noise ratios [51].
Streptavidin Purification Enhancements include bead titration to determine the optimal streptavidin bead-to-protein ratio, which prevents bead saturation and improves specific recovery [49]. Implementing stringent washing protocols with multiple buffer systems (e.g., SDS-containing buffers, deoxycholate buffers, and high-salt buffers) effectively removes nonspecifically bound proteins while retaining biotinylated targets [49].
Table 2: Technical Optimization Strategies for Background Reduction
| Workflow Stage | Optimization Strategy | Specific Protocol Recommendations | Expected Impact |
|---|---|---|---|
| Sample Preparation | Genetic depletion of endogenous biotinylated proteins | His-tagging of carboxylases for removal via Ni-based purification [2] | Reduces major source of biotin-dependent background |
| Labeling Reaction | Spatiotemporal control of enzyme activity | iAPEX system with DAAO for local H2O2 production [11] | Minimizes global activation of endogenous peroxidases |
| Protein Capture | Bead titration optimization | Fluorescence-based assessment of biotin capture efficiency [49] | Prevents bead saturation and improves specific recovery |
| Peptide Identification | Biotinylation site mapping | Acidic organo-aqueous denaturation buffer for peptide elution [33] | Direct identification of biotinylation sites eliminates ambiguity |
Advanced Methodologies: Peptide-Level Enrichment and Emerging Platforms
Biotinylation Site Mapping for Enhanced Specificity
Conventional PL workflows enrich biotinylated proteins at the protein level before tryptic digestion and mass spectrometry analysis. However, this approach co-purifies non-biotinylated proteins that associate with biotinylated targets, potentially yielding false positives [33] [2]. Peptide-level enrichment represents a significant advancement that directly identifies biotinylation sites, providing unambiguous evidence that proteins were genuinely labeled in situ.
A recently developed "super-resolution proximity labeling" method demonstrates the power of this approach [33]. This protocol involves digesting proteins before streptavidin capture, specifically enriching biotinylated peptides, and eluting them with an acidic organo-aqueous denaturation buffer. When applied to mitochondrial matrix proteomics, this method achieved an 89% true positive rate compared to 78.8% with conventional ratiometric approaches, while eliminating the need for complex negative controls [33].
The key advantages of biotinylation site identification include:
- Unambiguous verification of direct biotinylation rather than association with biotinylated complexes
- Elimination of negative controls in membrane-enclosed compartments, simplifying experimental design
- Enhanced detection of low-abundance proteins typically masked in protein-level enrichment
- Membrane topology inference through identification of specifically labeled domains
Emerging Enzyme Platforms with Reduced Background
Innovative PL enzymes with fundamentally different chemistries offer promising alternatives for background reduction:
LaccID is a recently engineered multicopper oxidase that uses O2 instead of toxic H2O2 for labeling [51]. Developed through 11 rounds of directed evolution from an ancestral fungal laccase, LaccID catalyzes one-electron oxidation of aromatic substrates and shows selective activity at the plasma membrane. Although its current catalytic efficiency is lower than HRP or APEX2, LaccID's different mechanism of action avoids background from endogenous peroxidases and functions without exogenous H2O2 [51].
iAPEX (in situ APEX activation) combines APEX2 with D-amino acid oxidase (DAAO) to create an enzyme cascade that locally generates H2O2 [11]. This system eliminates the need for external H2O2 addition, reducing both toxicity and background from endogenous peroxidases. The iAPEX system has successfully mapped proteomes in cell types previously inaccessible to conventional APEX labeling and has shown promise for in vivo applications in Xenopus laevis [11].
Experimental Workflows and Technical Implementation
Recommended Workflow for Background Minimization
The following diagram illustrates an optimized experimental workflow that incorporates critical steps for background reduction across the entire PL pipeline:
The Scientist's Toolkit: Essential Reagents and Methods
Table 3: Key Research Reagent Solutions for Background Reduction
| Reagent/Method | Specific Function | Application Notes |
|---|---|---|
| TurboID | Engineered biotin ligase for rapid labeling | Balance high efficiency with potential background from endogenous biotin; optimize concentration (50-500 μM) and time (10 min+) [3] [2] |
| APEX2 | Engineered peroxidase for ultrafast labeling | Use minimal H2O2 exposure (1 min); beware of endogenous peroxidases in some cell types [11] [2] |
| iAPEX System | Two-enzyme cascade (APEX2 + DAAO) | Eliminates exogenous H2O2; requires D-amino acids as substrates; reduces background in sensitive systems [11] |
| Streptavidin Magnetic Beads | Capture of biotinylated proteins | Titrate bead:protein ratio; use stringent washing buffers (SDS, deoxycholate, high-salt) [49] |
| Acidic Organo-aqueous Buffer | Elution of biotinylated peptides | Enables peptide-level enrichment; improves specificity by direct biotinylation site mapping [33] |
| D-Biotin | Substrate for biotin ligases | Use high-purity grade; optimize concentration for specific enzyme and cell type [3] |
| Biotin-Phenol/Biotin-Tyramide | Substrate for peroxidases | Membrane permeability can limit effectiveness; consider biotin-methoxyphenol for LaccID [51] |
The evolving landscape of proximity labeling technologies offers researchers multiple pathways to address the persistent challenge of background signals. The most effective approach involves matching the PL platform to the specific biological context while implementing appropriate controls and optimizations at each workflow stage. For membrane-enclosed compartments, emerging peptide-level enrichment methods provide unprecedented specificity by directly identifying biotinylation sites. For challenging cellular environments with high endogenous peroxidase activity, enzyme cascade systems like iAPEX offer a promising solution. As the PL field continues to mature, the strategic integration of these background reduction methods will enable researchers to extract clearer biological insights from increasingly complex experimental systems.
Proximity labeling (PL) coupled with mass spectrometry (MS) has revolutionized the study of subcellular proteomes and protein-protein interactions in living cells. However, after the initial biotinylation reaction, researchers face a critical methodological decision: whether to enrich and identify labeled proteins at the protein level or the peptide level. This choice fundamentally impacts the specificity, depth, and biological accuracy of the resulting interactome data. Protein-level enrichment, the conventional approach, involves capturing biotinylated proteins on streptavidin beads followed by on-bead digestion and identification of all resulting peptides. In contrast, peptide-level enrichment (also called biotinylation site mapping) involves digesting proteins first, then specifically enriching biotinylated peptides for liquid chromatography–tandem mass spectrometry (LC–MS/MS) analysis [33]. This guide provides an objective comparison of these two strategies, empowering researchers to select the optimal approach for their biological questions.
Fundamental Principles and Technical Workflows
Protein-Level Enrichment: The Conventional Approach
The protein-level enrichment workflow begins with cell lysis after the proximity labeling reaction, followed by incubation with streptavidin-coated beads to capture biotinylated proteins. After stringent washing, the captured proteins are digested on-bead with trypsin. The resulting peptides—both biotinylated and non-biotinylated from the same proteins—are then eluted and analyzed by LC–MS/MS [18] [33]. Identification relies on comparing protein abundance between experimental samples and negative controls (e.g., no enzyme expression or no H~2~O~2~/biotin treatment), typically using quantitative proteomic methods like tandem mass tag (TMT) labeling or label-free quantification [47]. Since most identified peptides lack the biotin modification itself, researchers must employ statistical filtering and ratiometric comparisons against controls to distinguish true proximal proteins from background [18].
Peptide-Level Enrichment: A Site-Specific Strategy
Peptide-level enrichment, also referred to as biotinylation site identification or mapping, reverses the order of operations. After cell lysis, the entire protein mixture is digested with trypsin. The resulting peptide pool is then incubated with streptavidin beads to specifically capture only those peptides containing the biotin modification. Following washing, the tightly bound biotinylated peptides are eluted under denaturing conditions and analyzed by LC–MS/MS [33]. This approach directly identifies the specific amino acid residues that were biotinylated, providing unequivocal evidence that a protein was within the labeling radius of the PL enzyme. The method significantly reduces background interference from non-biotinylated peptides and streptavidin, which often dominate samples in protein-level approaches [33].
Table 1: Core Workflow Comparison
| Step | Protein-Level Enrichment | Peptide-Level Enrichment |
|---|---|---|
| 1. Initial Processing | Cell lysis | Cell lysis, then protein precipitation to remove excess biotin [33] |
| 2. Digestion | On-bead tryptic digestion after enrichment [33] | In-solution denaturation and digestion with trypsin [33] |
| 3. Enrichment Target | Biotinylated proteins (streptavidin beads) [18] | Biotinylated peptides (streptavidin beads) [33] |
| 4. Elution | Mild conditions, often with Laemmli buffer or detergent [33] | Harsh conditions (e.g., acidic organo-aqueous denaturation buffer) [33] |
| 5. MS Identification | All peptides from enriched proteins | Only biotin-modified peptides |
The following workflow diagrams illustrate the key procedural differences between these two strategies:
Performance Comparison: Experimental Data and Metrics
Specificity and Background Reduction
Direct comparative studies demonstrate that peptide-level enrichment significantly outperforms protein-level approaches in specificity and true positive identification. In a systematic analysis of the mitochondrial matrix proteome using APEX2, peptide-level enrichment identified 449 true positive (TP) proteins out of 509 total identifications (88.2% TP rate). In contrast, protein-level enrichment with ratiometric control identified 471 TP out of 598 total identifications (78.8% TP rate) [33]. More notably, the quantitative composition of true positives within the final dataset was dramatically higher for peptide-level enrichment (89%) compared to protein-level approaches (24-36%) [33].
The background contamination from non-specifically bound proteins and streptavidin is substantially reduced in peptide-level workflows. The streptavidin signal in peptide-level analyses can be remarkably low, demonstrating superior cleanup during sample preparation [33]. This reduction in background interference translates to more reliable datasets and decreased analytical ambiguity.
Sensitivity and Depth of Identification
Recent methodological improvements have made peptide-level enrichment highly competitive in sensitivity. A novel optimized protocol for biotinylation site identification achieved a 2-fold increase in biotinylated peptide spectrum matches compared to the previous Spot-ID method and a 1.6-fold increase with 50% shorter LC–MS/MS gradient time compared to anti-biotin antibody approaches [33]. The enrichment efficiency for biotinylated peptides reached 89% with high inter-replicate reproducibility [33].
For membrane protein topology studies, peptide-level enrichment offers unique advantages by directly identifying biotinylation sites on specific domains, enabling researchers to infer membrane orientation and identify low-abundance membrane proteins that might be masked in protein-level approaches [18].
Table 2: Quantitative Performance Comparison (Mitochondrial Matrix Proteome)
| Performance Metric | Protein-Level Enrichment | Peptide-Level Enrichment |
|---|---|---|
| Total Identifications | 598 proteins [33] | 509 proteins [33] |
| True Positives (TP) | 471 proteins [33] | 449 proteins [33] |
| True Positive Rate | 78.8% [33] | 88.2% [33] |
| TP Quantitative Composition | 24-36% [33] | 89% [33] |
| Background Interference | High (streptavidin, non-specific binders) [33] | Significantly Reduced [33] |
| Experimental Design | Requires negative controls [18] | Can be performed without controls [33] |
Experimental Protocols and Methodological Details
Detailed Protocol: Protein-Level Enrichment
Cell Lysis and Capture:
- Lyse cells in RIPA buffer (or similar) containing protease inhibitors [17]
- Clarify lysate by centrifugation at 16,000 × g for 15 minutes
- Incubate supernatant with streptavidin-coated magnetic beads for 1-2 hours at 4°C with rotation [47]
- Recommended bead-to-lysate ratio: 50 μL bead slurry per 1-2 mg total protein
Stringent Washing:
- Wash sequentially with: (1) RIPA buffer, (2) 1M KCl, (3) 0.1M Na~2~CO~3~, (4) 2M urea in 10mM Tris-HCl (pH 8.0), and (5) 1X PBS [47]
- Perform each wash with 1 mL buffer per 50 μL bead slurry, incubating 5 minutes with rotation
On-Bead Digestion:
- Resuspend beads in 50mM ammonium bicarbonate (pH 8.0)
- Add trypsin at 1:50 (w/w) enzyme-to-protein ratio
- Digest overnight at 37°C with shaking
- Acidify with trifluoroacetic acid (0.5% final concentration) and collect supernatant [47]
Detailed Protocol: Optimized Peptide-Level Enrichment
Protein Digestion:
- Precipitate proteins to remove excess biotin using cold acetone or TCA [33]
- Resuspend protein pellet in 8M urea buffer for denaturation [33]
- Reduce with 5mM dithiothreitol (37°C, 45 minutes) and alkylate with 10mM iodoacetamide (room temperature, 30 minutes in dark)
- Dilute urea concentration to <1.5M with 50mM ammonium bicarbonate
- Digest with trypsin (1:50 w/w) overnight at 37°C [33]
Biotinylated Peptide Enrichment:
- Incubate digested peptides with streptavidin beads (2-4 hours, room temperature) [33]
- Wash stringently with: (1) 1% trifluoroacetic acid (TFA), (2) 50mM Tris-HCl (pH 7.5), 0.1% SDS, (3) 8M urea in 50mM Tris-HCl (pH 7.5), (4) 1M NaCl, and (5) 50mM ammonium bicarbonate [33]
Elution and Analysis:
- Elute biotinylated peptides using acidic organo-aqueous denaturation buffer [33]
- Dry eluents and resuspend in 25mM ammonium bicarbonate for LC–MS/MS analysis
- No additional desalting is required [33]
Research Reagent Solutions: Essential Materials for Implementation
Table 3: Key Reagents for Enrichment Strategies
| Reagent | Function | Example Use |
|---|---|---|
| Streptavidin Magnetic Beads | Captures biotinylated proteins/peptides | Used in both enrichment strategies [18] [33] |
| Trypsin, Sequencing Grade | Proteolytic digestion | Essential for both methods [33] |
| Desthiobiotin-Phenol (DBP) | APEX2 substrate (LC–MS friendly) | Alternative to biotin-phenol for improved biotinylated peptide analysis [33] |
| Acidic Organo-Aqueous Denaturation Buffer | Elutes biotinylated peptides | Critical for high-yield peptide-level enrichment [33] |
| Tandem Mass Tag (TMT) Reagents | Multiplexed quantitative proteomics | Enables ratiometric comparison in protein-level enrichment [47] |
| TurboID or APEX2 Enzymes | Proximity labeling catalysts | Generate the biotinylation signal for downstream analysis [47] |
Strategic Selection Guidelines and Applications
When to Choose Protein-Level Enrichment
Protein-level enrichment remains preferable in these scenarios:
- Pilot Studies: When initially characterizing a new subcellular compartment or protein complex
- Low-Input Samples: The method can be more forgiving with limited starting material
- Laboratories with Established Protocols: When infrastructure for protein-level workflows already exists
- Comparative Analyses: When quantitative comparisons between multiple conditions are needed using TMT or SILAC labeling [47]
When to Choose Peptide-Level Enrichment
Peptide-level enrichment is strongly recommended for:
- High-Confidence Interactome Mapping: When false positives are a major concern and maximal specificity is required [33]
- Membrane Protein Studies: When investigating membrane topology or low-abundance membrane proteins [18]
- Structured Ill-Defined Compartments: When labeling open cellular spaces rather than membrane-enclosed organelles [33]
- Control-Free Experimental Designs: The high specificity enables studies without negative controls, simplifying design [33]
- Advanced Applications: Such as mapping protein interfaces or precise localization determinants
The following decision tree provides a strategic framework for selecting the appropriate enrichment strategy:
The choice between protein-level and peptide-level enrichment in proximity labeling experiments represents a fundamental tradeoff between experimental convenience and data specificity. While protein-level enrichment offers established protocols and may be more accessible for preliminary studies, peptide-level enrichment provides superior specificity, reduced background, and direct evidence of biotinylation through site identification. As methodological improvements continue to enhance the sensitivity and accessibility of peptide-level approaches, this strategy is increasingly becoming the gold standard for high-confidence interactome mapping. Researchers should select their enrichment strategy based on their specific biological questions, required confidence levels, and technical capabilities, using the comparative data and guidelines presented herein to inform their experimental design.
In the field of intracellular research, particularly with the rise of proximity-labeling (PL) techniques for mapping protein interactions, the choice of expression system is a critical determinant of experimental success. The fundamental goal is to study the protein of interest (POI) in a context that most accurately reflects its native expression, localization, and function. Two predominant strategies for this are the generation of stable cell lines and endogenous tagging [52] [53] [54]. Stable cell lines involve the integration of a transgene—a fusion of the POI and a tag or enzyme (e.g., for PL)—into a consistent genomic location, allowing for persistent expression. In contrast, endogenous tagging uses gene-editing technologies to insert a tag directly into the native genomic locus of the POI, ensuring its expression is controlled by its endogenous regulatory elements [53] [55] [54]. This guide provides an objective comparison of these two systems, framing the discussion within the practical requirements of researchers employing modern intracellular tagging and proximity-labeling methodologies.
Key Technological Concepts and Definitions
To understand the comparison, it is essential to first define the core technologies and reagents involved.
Stable Cell Lines are typically created using systems like Flp-In [52]. This technology relies on site-specific recombination between an FRT site in the host cell's genome and an FRT site on a transfected plasmid. This allows for the targeted integration of a single transgene copy at a defined genomic location, enabling consistent, reproducible expression across a polyclonal population [52]. Inducible promoters (e.g., Tet-On) can be used to control the timing and level of protein expression, mitigating toxicity associated with constitutive overexpression [52].
Endogenous Tagging is powered by CRISPR/Cas9 gene editing, which creates a double-strand break at a specific site in the genome directed by a guide RNA (gRNA) [53] [54]. The cell's repair machinery then uses a provided donor DNA template—often a single-stranded oligodeoxynucleotide (ssODN) for small tags—to incorporate the tag (e.g., HiBiT, mNG211, FLAG) via homology-directed repair (HDR), resulting in a fusion protein expressed from the native promoter [53] [55] [54].
Proximity-Labeling (PL) Enzymes are a key application for these expression systems. PL enzymes like TurboID and APEX2 are fused to a POI. Upon addition of a substrate (biotin for TurboID; biotin-phenol and H₂O₂ for APEX2), they catalyze the biotinylation of proximal proteins, which can then be captured and identified via mass spectrometry [56] [57] [58]. The choice of expression system directly impacts the fidelity of the resulting interactome.
Research Reagent Solutions
The table below details essential reagents and their functions for implementing these technologies.
| Reagent Category | Specific Examples | Function in Experimental Workflow |
|---|---|---|
| Tagging Systems | LAP-tag (EGFP-TEV-S-peptide) [52], HiBiT [53], FLAG epitope [55], split mNeonGreen [54] | Provides a handle for protein detection, purification, or live-cell imaging. |
| Cloning & Vector Systems | Gateway-compatible vectors (e.g., pgLAP1) [52], AAV-based targeting vectors [55] | Facilitates high-throughput and efficient movement of gene sequences into expression constructs. |
| Cell Line Engineering Tools | Flp-In T-REx cell lines [52], engineered iPSCs expressing mNG21-10 [54] | Provides standardized, genetically defined parental cells for consistent stable line generation or endogenous tagging. |
| Proximity Labeling Enzymes | TurboID, miniTurbo, APEX2 [56] [58] | Genetically encoded enzymes that label nearby biomolecules for interactome mapping. |
| Labeling Substrates | Biotin (for BioID/TurboID), Biotin-phenol & H₂O₂ (for APEX/APEX2) [56] [57] [58] | Enzymatic substrates converted into reactive intermediates that covalently tag proximal proteins. |
Systematic Comparison of Methodologies
The following tables provide a detailed, side-by-side comparison of stable cell lines and endogenous tagging across critical performance parameters.
Table 1: Direct Comparison of Key Characteristics
| Feature | Stable Cell Lines (e.g., Flp-In) | Endogenous Tagging (e.g., CRISPR/Cas9) |
|---|---|---|
| Protein Expression Level | Non-physiological, often high and driven by a strong exogenous promoter (e.g., CMV) [53]. | Physiological, controlled by the native endogenous promoter [53] [54]. |
| Subcellular Localization | Risk of mislocalization due to overexpression and lack of native regulatory sequences [54]. | High fidelity, as all native regulatory elements are preserved [54]. |
| Temporal Control | Excellent with inducible systems (e.g., Tet-On), allowing precise control over expression timing [52]. | Limited to the natural timing of endogenous gene expression. |
| Technical Throughput | High for parallel generation once the parental line is established [52]. | Scalable with careful optimization, as demonstrated by large-scale tagging efforts [53] [54]. |
| Typical Integration Locus | Defined, transcriptionally active "safe harbor" site (e.g., Flp-In locus) [52]. | The native genomic locus of the POI. |
| Representative Experimental Timeline | ~2-3 weeks from cloning to polyclonal selection [52]. | ~4-8 weeks including clonal screening and validation [53] [54]. |
Table 2: Comparison of Experimental Advantages and Limitations
| Aspect | Stable Cell Lines | Endogenous Tagging |
|---|---|---|
| Key Advantages | • Avoids clonal variation via polyclonal populations [52].• Inducible expression circumvents toxicity [52].• Generally faster and more reliable generation. | • Eliminates overexpression artifacts [53] [54].• Preserves natural stoichiometry and interactions.• Reveals authentic protein dynamics and regulation. |
| Major Limitations | • Overexpression artifacts: mislocalization, non-native interactions [53] [54].• Disrupted native transcriptional regulation. | • Technically challenging; lower HDR efficiency than NHEJ [54].• Risk of incomplete tagging and clonal variation.• Potential for off-target editing effects. |
| Ideal Use Cases | • High-throughput interaction screens.• Studying proteins whose overexpression is toxic (with inducible systems) [52].• Expressing mutant forms or tagged proteins in a uniform background. | • Validating interactions discovered in overexpression screens.• Studying proteins in their native context is critical.• Live imaging of dynamic processes in "healthy" cell models like iPSCs [54]. |
Experimental Protocols and Workflows
Below are generalized protocols for generating stable cell lines and creating endogenously tagged cells, highlighting the key methodological differences.
Protocol for Generating Inducible Stable Cell Lines
This protocol is adapted from methods using the Flp-In T-REx system and LAP-tagging [52].
- Vector Construction: Clone the gene of interest (GOI), fused to your PL enzyme (e.g., TurboID), into a Gateway-compatible destination vector (e.g., pgLAP1) containing the inducible promoter and an FRT site [52].
- Cell Line Preparation: Culture Flp-In T-REx host cells, which stably express the Tet repressor and have a single integrated FRT site.
- Transfection: Co-transfect the purified GOI/pgLAP1 plasmid with a plasmid expressing the Flp recombinase (pOG44) into the host cells [52].
- Selection and Expansion: At 24 hours post-transfection, begin selection with the appropriate antibiotic (e.g., hygromycin). The Flp recombinase mediates the integration of the GOI plasmid into the FRT site, conferring antibiotic resistance. Expand the entire polyclonal population for experiments, bypassing the need for single-cell cloning [52].
- Induction and Validation: To express the PL fusion protein, add doxycycline to the culture medium. Validate the expression and functionality of the fusion protein through Western blot, imaging, and PL activity assays.
Protocol for Endogenous Tagging with CRISPR/Cas9
This protocol is based on methods using CRISPR/HiBiT tagging and the split mNeonGreen system in human cells [53] [54].
- gRNA and Donor Design: Design gRNAs to create a double-strand break as close as possible to the stop codon (for C-terminal tagging) of the endogenous gene. Synthesize an ssODN donor template containing the tag (e.g., HiBiT, mNG211) flanked by homology arms (typically 50-80 bp) that match the sequences around the cut site [53]. The donor should be designed to disrupt the PAM site to prevent re-cutting.
- Delivery of Editing Components: Electroporate or transfect cells with the ribonucleoprotein (RNP) complex—consisting of recombinant Cas9 protein and the synthetic gRNA—along with the ssODN donor template [53]. Using an RNP complex can increase efficiency and reduce off-target effects.
- Clonal Isolation and Screening: After delivery, culture cells and isolate single clones by fluorescence-activated cell sorting (FACS) or limiting dilution. Screen the clones for correct integration using a combination of methods:
- Validation and Expansion: Expand positive clones and perform further validation, such as Western blotting to confirm the expected molecular weight of the tagged protein and functional assays to ensure the tag does not disrupt protein activity.
The following diagram illustrates the core logical and technical differences between the two workflows.
The choice between stable cell lines and endogenous tagging is not a matter of which is universally superior, but rather which is most appropriate for the specific research question and context.
For exploratory, high-throughput interactome mapping using proximity labeling, stable cell lines offer a powerful and pragmatic solution. The ability to use inducible systems is a significant advantage for studying proteins essential for cell viability [52]. The standardized Flp-In protocol and the generation of polyclonal populations make this approach highly reproducible and scalable for screening dozens to hundreds of proteins [52]. However, data generated from these systems must be interpreted with the caveat that overexpression may reveal interactions that are not physiologically relevant or may miss interactions that require proper stoichiometry [53] [54].
In contrast, endogenous tagging is the unequivocal method for validating biological findings and studying protein function under physiological conditions. It is indispensable for research in sensitive models like human induced pluripotent stem cells (iPSCs), where authentic cellular processes are paramount [54]. As the field moves towards more accurate and nuanced models of cellular biology, particularly for drug discovery and understanding disease mechanisms, endogenous tagging provides a level of biological fidelity that overexpression systems cannot match. The development of highly efficient small tags like HiBiT and split fluorescent proteins has made this approach increasingly accessible and scalable [53] [54].
In conclusion, stable cell lines and endogenous tagging are complementary tools in the modern researcher's arsenal. A robust research strategy may often begin with a screen in a well-controlled stable cell line system, with critical findings subsequently validated in an endogenously tagged model. This two-tiered approach leverages the strengths of both methodologies to generate discoveries that are both broad-reaching and biologically profound.
Proximity labeling (PL) has emerged as a transformative technology for studying protein-protein interactions, subcellular proteomes, and spatial organization of proteins within their native cellular environments. Unlike traditional methods like affinity purification or yeast two-hybrid systems, PL enables the covalent tagging of neighboring proteins in living cells, preserving transient interactions and capturing molecular relationships that would be lost during cell lysis [28] [8]. This technique relies on engineered enzymes that generate reactive molecules to biotinylate proteins within a limited radius, after which the tagged proteins can be purified under stringent conditions and identified via liquid chromatography-mass spectrometry (LC-MS) [21].
The critical advantage of PL lies in its ability to capture weak, transient, and spatially restricted interactions while minimizing false positives that commonly arise from traditional purification methods [28]. As proteomic studies increasingly focus on dynamic cellular processes and subtle regulatory mechanisms, managing experimental variation from the initial labeling reaction through LC-MS analysis becomes paramount for generating reproducible, biologically relevant data. This guide provides a comprehensive comparison of proximity-labeling enzymes and their optimal implementation for intracellular tagging research.
Comparative Analysis of Proximity-Labeling Enzymes
Enzyme Classifications and Properties
Proximity-labeling enzymes primarily fall into two major categories: peroxidases and biotin ligases, each with distinct mechanisms, advantages, and limitations [28] [21]. Peroxidases like APEX and APEX2 utilize hydrogen peroxide (H₂O₂) to oxidize biotin-phenol substrates into phenoxyl radicals that covalently tag electron-rich amino acids on nearby proteins [28]. In contrast, biotin ligases such as BioID and TurboID use ATP to activate biotin to biotin-AMP, which then diffuses to label lysine residues on proximal proteins [28] [21].
Table 1: Comparison of Major Proximity-Labeling Enzymes
| Enzyme | Class | Size (kDa) | Labeling Radius | Labeling Time | Activation Requirement | Key Advantages | Major Limitations |
|---|---|---|---|---|---|---|---|
| BioID | Biotin Ligase | ~35 | ~10 nm | 18-24 hours | ATP/Biotin | Low background, well-established | Slow kinetics, not ideal for dynamic processes |
| TurboID | Biotin Ligase | ~35 | ~10-35 nm | 10 min - 2 hours | ATP/Biotin | Extremely fast labeling, high sensitivity | Cellular toxicity at high expression, broader labeling radius |
| miniTurbo | Biotin Ligase | ~28 | ~10 nm | 10 min - 2 hours | ATP/Biotin | Fast labeling with reduced background | Lower activity than TurboID |
| APEX/APEX2 | Peroxidase | ~28 | <20 nm | 1 minute | H₂O₂/Biotin-phenol | Exceptional temporal resolution, small labeling radius | H₂O₂ toxicity, poor membrane permeability of biotin-phenol |
| HRP | Peroxidase | ~44 | <20 nm | 1-5 minutes | H₂O₂/Various substrates | High activity with various substrates | Limited to oxidizing environments (e.g., secretory pathway) |
Experimental Considerations for Enzyme Selection
Selecting the appropriate PL enzyme requires careful consideration of experimental goals and biological constraints. For dynamic processes requiring high temporal resolution, APEX2 (1-minute labeling) or TurboID (10-minute labeling) are preferable despite potential toxicity concerns [28]. For localization studies where precise spatial information is critical, enzymes with smaller labeling radii like APEX2 (<20 nm) or BioID (~10 nm) provide higher resolution [28] [21].
Recent engineering efforts have produced specialized variants addressing specific experimental needs. Split-enzyme systems (Split-TurboID, Split-BioID) enable detection of specific protein-protein interactions by reconstituting only when two fragments are brought together [21]. Environment-activated enzymes like Cal-ID (calcium-sensitive) and LOV-TurboID (light-activated) provide spatial and temporal control over labeling [6] [19]. For example, Cal-ID biotinylates nearby proteins in response to elevated Ca²⁺ concentrations, allowing biochemical recording of calcium signaling and neuronal activity [19].
Experimental Protocols and Methodologies
Standardized Workflow for Proximity Labeling
Implementing consistent experimental protocols is essential for minimizing variation in PL studies. The following workflow represents a generalized procedure applicable to most PL enzymes, with specific modifications noted for different enzyme classes:
Construct Design and Expression: Fuse the PL enzyme to your protein of interest using appropriate linkers (typically 15-20 amino acids) to minimize steric hindrance. Consider subcellular localization signals if targeting specific organelles. For biotin ligases, use lower expression levels to reduce toxicity; for peroxidases, ensure compatibility with the cellular environment [28] [21].
Labeling Reaction Optimization:
- For biotin ligases: Add biotin to culture media at optimized concentrations (typically 50-500 μM). The labeling duration varies from 10 minutes (TurboID) to 24 hours (BioID) based on enzyme kinetics [28].
- For peroxidases: Add biotin-phenol (500 μM) followed by hydrogen peroxide (1 mM) for exactly 1 minute before rapid quenching with scavenger solutions (e.g., Trolox, sodium ascorbate) [28].
Cell Lysis and Protein Extraction: Use RIPA buffer containing protease inhibitors and 0.1% SDS. Include 1-2% SDS for complete solubilization of membrane proteins, with subsequent dilution to 0.1-0.2% for compatibility with streptavidin beads [21].
Streptavidin Purification: Incubate lysates with streptavidin-coated beads for 1-2 hours at room temperature. Perform stringent washing with sequential buffers: (1) RIPA with 0.1% SDS, (2) high-salt buffer (1 M KCl), (3) carbonate buffer (100 mM Na₂CO₃), and (4) 50 mM Tris pH 7.4 [21].
On-Bead Digestion and LC-MS Preparation: Reduce (5 mM DTT) and alkylate (15 mM iodoacetamide) proteins on beads, followed by tryptic digestion overnight. Desalt peptides using C18 StageTips before LC-MS analysis [59].
Diagram 1: Proximity Labeling Experimental Workflow
Quality Control and Validation Methods
Rigorous quality control measures are essential throughout the PL workflow. Implement these validation steps:
Labeling Efficiency Assessment: Perform western blotting with streptavidin-HRP to visualize biotinylation patterns before purification. Successful labeling shows strong, specific biotinylation in experimental versus control samples [21].
Specificity Verification: Include critical negative controls: (1) catalytically dead enzyme mutants, (2) samples without substrate addition, and (3) untargeted enzymes (e.g., cytosolic expression without fusion partner) [28].
Quantitative MS Controls: Use stable isotope labeling (SILAC, TMT) or label-free quantification with internal standards to distinguish specific interactions from background [59]. Incorporate cross-linking validation for critical interactions when possible.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Research Reagent Solutions for Proximity Labeling
| Reagent Category | Specific Examples | Function & Importance | Optimization Tips |
|---|---|---|---|
| PL Enzymes | TurboID, APEX2, BioID2 | Covalently tags proximal proteins with biotin | Select based on temporal needs: TurboID for speed, BioID for precision |
| Biotin Substrates | Biotin (ligases), Biotin-phenol (peroxidases) | Enzyme-specific substrates for radical generation | Use fresh stocks; optimize concentration to balance signal vs. background |
| Activation Reagents | H₂O₂ (peroxidases), None required (ligases) | Triggers radical formation in peroxidases | Precisely control H₂O₂ concentration (typically 1 mM) and time to minimize toxicity |
| Quenchers | Trolox, sodium ascorbate, catalase | Stops labeling reaction (peroxidases) | Add immediately after H₂O₂ incubation; use fresh solutions |
| Purification Matrix | Streptavidin-coated beads | Captures biotinylated proteins | Use high-capacity, low-background beads; avoid overloading |
| Lysis Buffers | RIPA with SDS, protease inhibitors | Extracts proteins while preserving modifications | Include 0.1-0.2% SDS for membrane proteins; optimize for your system |
| MS Standards | TMT, SILAC amino acids | Enables quantitative comparison across samples | Essential for distinguishing specific interactions from background |
Managing Variation in LC-MS Analysis
Quantitative Proteomics Approaches
LC-MS analysis of PL samples presents unique challenges due to the complex nature of biotinylated protein mixtures. Several quantitative approaches help manage experimental variation:
Stable Isotope Labeling methods like SILAC (stable isotope labeling with amino acids in cell culture) and tandem mass tags (TMT) enable multiplexed analysis of multiple conditions within the same MS run, minimizing technical variation [59]. SILAC incorporates heavy isotopes metabolically during cell culture, while TMT uses isobaric tags chemically added to peptides before LC-MS. For PL studies, these methods are particularly valuable for distinguishing specific protein interactions from background binders when comparing bait-dependent versus control samples [59].
Label-Free Quantification (LFQ) offers an alternative without chemical labeling, using peptide signal intensities or spectral counting across multiple runs. While more accessible, LFQ requires careful normalization and additional replicates to account for run-to-run variability [59]. Advanced algorithms like MaxLFQ have improved the reliability of this approach for PL data.
Data Processing and Normalization Strategies
Proper data processing is crucial for accurate interpretation of PL-MS experiments. Implement these strategies:
Background Subtraction: Use multiple negative controls (e.g., untargeted enzyme, no substrate, dead enzyme) to create a background model for subtraction. The SAINT (Significance Analysis of INTeractome) algorithm is specifically designed for this purpose in interaction proteomics [21].
Normalization Approaches: Apply variance-stabilizing normalization methods like vsn or quantile normalization to address technical variation. Use total peptide amount or proteomic housekeeping proteins for normalization reference.
Statistical Analysis: Employ false discovery rate (FDR) correction (typically 1-5%) using the Benjamini-Hochberg method. Implement intensity-based filtering to remove low-confidence identifications.
Diagram 2: LC-MS Data Processing with Quality Control
Emerging Technologies and Future Directions
The field of proximity labeling continues to evolve with new enzymes and methods addressing current limitations. Recent developments include:
H₂O₂-Independent Enzymes such as bacterial tyrosinase (BmTyr) and engineered fungal laccase (LaccID) utilize molecular oxygen instead of potentially toxic H₂O₂, improving cellular compatibility [19]. BmTyr enables rapid (≤10 min) labeling with low background, while LaccID offers specific labeling of cell surface proteomes.
Cascade Labeling Systems combine multiple enzymes for enhanced spatial control. For example, the P2L system uses galactose oxidase to generate H₂O₂ specifically at glycosylation sites, which then activates HRP for localized labeling [19]. Similarly, singlet oxygen photosensitizing protein-3 can be coupled with APEX2 to create H₂O₂ in situ upon blue light illumination, enabling precise spatial control without exogenous H₂O₂ addition [19].
Endogenous Targeting Approaches leverage ligands, antibodies, or aptamers to direct PL enzymes to native proteins without genetic manipulation. The μMap platform conjugates iridium photocatalysts to antibodies for labeling antibody-binding targets and their neighbors [19]. Small molecule-PL enzyme conjugates enable mapping of endogenous neurotransmitter receptor proximal proteomes in live mouse brain, demonstrating the potential for in vivo applications [19].
These advancements continue to refine the spatial and temporal resolution of proximity labeling while expanding its applicability to challenging biological systems, including clinical samples and intact organisms. As these technologies mature, they will further reduce experimental variation and enhance the reproducibility of proximity-based proteomic studies.
Head-to-Head Comparison: Validating and Selecting the Optimal PL Enzyme
Proximity labeling (PL) has revolutionized the study of protein-protein interactions and subcellular proteomes by enabling the covalent tagging of proximal proteins within living cells. For researchers investigating intricate intracellular environments, selecting the appropriate labeling enzyme is crucial for experimental success. This guide provides a direct performance comparison of three widely used PL enzymes—TurboID, APEX2, and BioID2—drawing on recent experimental data and methodological studies to inform your choice for intracellular tagging research.
At a Glance: Core Characteristics and Performance Metrics
The following table summarizes the key characteristics and performance metrics of TurboID, APEX2, and BioID2, providing a foundation for their direct comparison.
Table 1: Direct Performance Comparison of Proximity Labeling Enzymes
| Feature | TurboID | APEX2 | BioID2 |
|---|---|---|---|
| Enzyme Origin | Mutant E. coli biotin ligase (BirA), engineered via directed evolution [18] [47] | Engineered ascorbate peroxidase (APX) [47] | Mutant E. coli biotin ligase (BirA), truncated and optimized [18] [60] |
| Primary Intracellular Use | General intracellular labeling [61] | General intracellular labeling [47] | General intracellular labeling [18] |
| Labeling Radius | ~10-20 nm (estimated) [60] | ~20 nm [60] | ~10 nm [60] |
| Labeling Time | Minutes to 1 hour [18] [60] | Seconds to Minutes [18] [60] | Several hours (often 12-18 hours) [18] [60] |
| Key Substrates | Biotin + ATP [61] | Biotin-phenol (or derivatives) + H₂O₂ [11] [47] | Biotin + ATP [18] |
| Cytotoxicity / Limitations | High catalytic activity may cause cell stress or non-specific background labeling; requires careful optimization of time and biotin concentration [18]. | Cytotoxic due to high H₂O₂ requirement; can induce oxidative stress, limiting some in vivo applications [11] [18]. | Long labeling times preclude capture of rapid dynamic interactions [18]. |
| Key Advantages | Extremely fast kinetics; easy to use (only requires biotin); compatible with a wide range of model organisms, including animals [18] [47]. | Ultra-fast labeling ideal for capturing highly transient interactions; can generate EM contrast; can label other biomolecules like RNA [18] [62] [47]. | Smaller size may reduce steric hindrance; well-established protocol [18] [60]. |
Experimental Workflows and Key Methodologies
The performance characteristics of each enzyme directly inform the design of experimental protocols. Below are detailed methodologies for key experiments cited in recent literature, which can serve as templates for your own research.
TurboID-Based Interactome Mapping
A comprehensive study of the PARP family utilized V5-TurboID fusion constructs in HEK293T cells to identify high-confidence protein interactors [61]. The protocol is highly standardized and reproducible.
Key Experimental Protocol [61]:
- Plasmid Transfection: Transfect cells with 15 µg of V5-TurboID-tagged bait plasmid using polyethylenimine (PEI).
- Expression & Labeling: Allow protein expression for 24-48 hours. For labeling, incubate cells with 50 µM biotin in culture media for 1 hour at 37°C.
- Cell Lysis: Wash cells with ice-cold PBS and lyse in SDS-Lysing buffer (1% SDS, 50 mM Tris-HCl pH 8) at 95°C for 15 minutes to ensure denaturation.
- Biotinylated Protein Enrichment: Dilute lysate and incubate with streptavidin magnetic beads overnight at room temperature.
- On-bead Digestion: Wash beads thoroughly. Elute and digest proteins using trypsin (1 µg/µL) with shaking at 37°C for 12-18 hours.
- Mass Spectrometry Analysis: Desalt peptides using C18 microcolumns and analyze via LC-MS/MS.
This workflow successfully identified 6,314 high-confidence interacting proteins, capturing transient interactions often missed by conventional methods [61].
APEX2 Labeling with Enhanced Specificity (iAPEX)
A major challenge for conventional APEX2 is non-specific background labeling from endogenous peroxidases activated by exogenous H₂O₂. The innovative iAPEX (in situ APEX activation) workflow overcomes this by generating H₂O₂ locally [11].
Key Experimental Protocol [11]:
- DAAO Co-expression: Co-express the bait-APEX2 fusion with a D-amino acid oxidase (DAAO), targeted to the same subcellular location (e.g., primary cilium).
- Enzymatic Cascade Activation: To initiate labeling, add the DAAO substrate (e.g., D-alanine) along with biotin-phenol.
- Localized H₂O₂ Production: DAAO oxidizes D-alanine, locally producing H₂O₂, which is immediately used by the nearby APEX2 to activate biotin-phenol.
- Downstream Processing: Proceed with standard cell lysis, streptavidin-based enrichment, and MS analysis.
This method eliminates the need for toxic, high-concentration H₂O₂ and drastically reduces non-specific background, enabling proteomic profiling in cell lines previously incompatible with APEX2 [11].
BioID2 for Synaptic Proteome Profiling
BioID2, a smaller and optimized variant of BioID, has been effectively applied in neuroscience to profile the synaptic proteome, a complex and dynamic cellular compartment [18] [60].
Key Experimental Protocol [18] [60]:
- Construct Design: Fuse the synaptic protein of interest (bait) with BioID2.
- Long-term Labeling: Express the construct in neurons and supplement the culture medium with biotin for several hours (typically 12-18 hours) to allow for biotinylation.
- Tissue Processing & Enrichment: Lyse cells or brain tissue and enrich biotinylated proteins using streptavidin or NeutrAvidin beads.
- Protein Identification: Identify captured proteins via LC-MS/MS.
This approach benefits from the smaller size of BioID2, which may reduce steric interference with the native function and localization of the synaptic bait protein [60].
Visual Guide to Enzyme Mechanisms and Workflows
The following diagrams illustrate the distinct catalytic mechanisms and experimental setups for each proximity labeling enzyme, highlighting their unique operational principles.
TurboID and BioID2 Catalytic Mechanism
TurboID/BioID2 Mechanism: Both enzymes use ATP to convert biotin into a reactive biotin-AMP intermediate, which is released and covalently attaches to lysine residues on nearby proteins. Their key difference lies in catalytic efficiency, with TurboID being vastly faster [18].
APEX2 Catalytic Mechanism
APEX2 Mechanism: APEX2 uses hydrogen peroxide (H₂O₂) to oxidize biotin-phenol into a highly reactive, short-lived phenoxyl radical that instantly labels tyrosine residues on proximal proteins [18] [47].
iAPEX Workflow for Specific Labeling
iAPEX Workflow: The iAPEX system co-localizes D-amino acid oxidase (DAAO) with APEX2. Adding a D-amino acid triggers local H₂O₂ production, which activates APEX2 specifically at the site of interest, minimizing background and toxicity [11].
The Scientist's Toolkit: Essential Research Reagents
Successful proximity labeling experiments require careful selection of reagents and controls. The following table details key solutions used in the featured protocols.
Table 2: Essential Research Reagent Solutions
| Reagent / Solution | Function / Description | Example Use Case |
|---|---|---|
| V5-TurboID Plasmid | Mammalian expression vector for generating N-terminal V5-TurboID fusion proteins with a standardized linker [61]. | Systematic interactome mapping of protein families under unified conditions [61]. |
| Streptavidin Magnetic Beads | High-affinity solid-phase matrix for purifying biotinylated proteins from complex cell lysates. | Standard enrichment step in all biotin-based PL protocols (TurboID, APEX2, BioID2) [61] [18]. |
| Desthiobiotin-Phenol (DBP) | A reversible, high-affinity APEX substrate. Allows for gentler elution of labeled proteins compared to biotin-phenol, potentially improving protein recovery [63]. | Used in quantitative, peptide-level APEX studies to enable efficient elution and mapping of biotinylation sites [63]. |
| D-Amino Acid Oxidase (DAAO) | Enzyme that oxidizes D-amino acids to locally produce H₂O₂, eliminating the need for exogenous H₂O₂ addition. | Core component of the iAPEX system to reduce toxicity and background in sensitive cell types [11]. |
| Isotope-Coded Phenol Probes (e.g., LDBP/HDBP) | Stable isotope-labeled versions of APEX substrates (e.g., Desthiobiotin-Phenol) for multiplexed, quantitative proteomics. | Enables duplexed super-resolution PL (ICAX) to compare protein distributions between two bait proteins [63]. |
TurboID, APEX2, and BioID2 are powerful tools that cater to different experimental priorities in intracellular proteomics. TurboID is the preferred choice for its ease of use, speed, and broad in vivo compatibility. APEX2 is unmatched for temporal resolution and capturing ultra-fast, transient interactions, with innovations like iAPEX mitigating its toxicity. BioID2 offers a smaller, well-characterized alternative to the original BioID but is limited by its slow kinetics.
Your ultimate choice should be guided by the specific biological question: the need for speed, compatibility with your model system, and the required spatial resolution. As the field evolves, new engineered enzymes and refined protocols will continue to expand the possibilities for mapping the intricate molecular landscape of the cell.
Quantitative proteomics is indispensable for understanding dynamic cellular processes, from signaling networks to protein interaction dynamics. Among the many techniques available, Stable Isotope Labeling by Amino Acids in Cell Culture (SILAC) and Tandem Mass Tags (TMT) represent two cornerstone strategies for mass spectrometry-based relative quantitation. A sophisticated advancement, known as ratiometric tagging, further combines these principles to achieve unprecedented spatial specificity in complex biological studies. This guide provides an objective comparison of these techniques, focusing on their performance characteristics, supported by experimental data, and frames them within the context of modern intracellular tagging research, particularly in conjunction with proximity-labeling enzymes.
Technical Comparison of SILAC and TMT
The choice between SILAC and TMT involves trade-offs between quantification accuracy, multiplexing capability, and experimental applicability. The table below summarizes their core characteristics based on empirical studies.
Table 1: Core Characteristics of SILAC and TMT
| Feature | SILAC (Stable Isotope Labeling by Amino Acids in Cell Culture) | TMT (Tandem Mass Tags) |
|---|---|---|
| Labeling Type | Metabolic labeling in vivo [64] [65] | Chemical labeling (isobaric tags) on peptides in vitro [65] [66] |
| Principle | Incorporation of "light" or "heavy" amino acids during cell culture [64] | Peptides from different samples labeled with unique isobaric tags; reporter ions released during MS/MS [65] [66] |
| Multiplexing Capacity | Typically 2-3 plex (Standard); Up to 4 plex with NeuCode [64] | High-plex: Up to 16-18 samples simultaneously [65] [66] |
| Quantification Level | MS-level (precursor intensity) [65] | MS2- or MS3-level (reporter ions) [67] [66] |
| Key Advantage | High quantitative accuracy; minimal chemical artifacts; ideal for dynamic process studies [68] [65] | High multiplexing reduces run-to-run variation; high throughput for complex sample sets [65] [66] |
| Key Limitation | Limited to cell culture systems; requires multiple cell divisions for full incorporation [65] | Susceptible to "ratio compression" due to co-isolation of peptides [65] [66] |
| Ideal Use Case | Studying dynamic processes (e.g., protein turnover, signaling) in cell culture [67] [65] | High-throughput screening of multiple conditions (e.g., time courses, drug doses) [65] |
Performance Data from Direct Comparative Studies
A systematic comparison of label-free, SILAC, and TMT techniques provides critical performance data to guide method selection.
Table 2: Experimental Performance Data from a Systematic Comparison [68]
| Performance Metric | Label-Free | SILAC | TMT |
|---|---|---|---|
| Protein/Coverage | Superior coverage [68] | Intermediate coverage [68] | Lowest coverage, most missing values [68] |
| Technical Variability (Proteome) | Higher variability [68] | Lowest variability (Highest precision) [68] | Lower variability than label-free [68] |
| Technical Variability (Phosphoproteome) | Higher variability [68] | Outstanding performance and precision [68] | Lower variability than label-free [68] |
| Note on TMT | - | - | Performance decreases if experimental replicates are distributed across multiple TMT plexes [68] |
This study on the EGFR signaling network in colorectal cancer cells concluded that SILAC showed the highest precision and outstanding performance for quantification of phosphosites, making it the preferred method for analyzing cellular signaling in cell culture models [68].
Workflow and Signaling Pathway Diagrams
Understanding the workflows of these techniques and the signaling pathways they can elucidate is crucial for experimental design. The following diagrams outline the core procedures and a key biological application.
SILAC and TMT Experimental Workflows
EGFR Signaling Pathway and Proteomic Insight
Quantitative proteomics, particularly phosphoproteomics, is vital for dissecting signaling pathways. The diagram below illustrates the EGFR signaling network and the adaptive mechanisms revealed by the cited study [68].
Advanced Application: Ratiometric Tagging for Spatial Proteomics
A powerful application of quantitative proteomic strategies is their integration with proximity labeling (PL) to map subcellular proteomes with high spatial resolution.
The Challenge and The Ratiometric Solution
Traditional APEX labeling in compartments like the mitochondrial intermembrane space (IMS) can suffer from high background because reactive biotin-phenoxyl radicals can escape and label nearby cytosolic proteins [69]. To address this, researchers developed a SILAC-based ratiometric tagging strategy [69]. This method, inspired by ratiometric fluorescent reporters, measures the ratio of a protein's biotinylation by two different APEX constructs: one targeted to the compartment of interest (e.g., IMS-APEX) and one targeted to an adjacent compartment (e.g., cytosolic APEX) [69].
Ratiometric Tagging Experimental Protocol
Step 1: Cell Line Preparation and Labeling
- Generate cell lines expressing the PL enzyme (e.g., APEX2) targeted to your compartment of interest (e.g., IMS-APEX) and a control compartment (e.g., cytosolic APEX) [69].
- Culture the IMS-APEX cell line in SILAC "heavy" media and the cytosolic-APEX cell line in SILAC "light" media [69].
Step 2: Parallel Proximity Labeling
- Perform APEX-mediated biotinylation simultaneously on both cell populations by adding biotin-phenol and hydrogen peroxide for a short pulse (e.g., 1 minute) [69] [28].
- Immediately after labeling, mix the "heavy" and "light" cells in a 1:1 ratio and lyse them [69].
Step 3: Affinity Purification and Mass Spectrometry
- Enrich biotinylated proteins using streptavidin-coated magnetic beads under stringent conditions [69] [8].
- Process the enriched proteins for LC-MS/MS analysis [69].
Step 4: Data Analysis and Hit Selection
- For each identified protein, calculate two ratios:
- Biotinylation Enrichment: The degree of enrichment in the streptavidin pull-down versus the input lysate.
- Spatial Ratio (Ratiometric): The SILAC H/L ratio, which reflects the relative efficiency of biotinylation by IMS-APEX versus cytosolic APEX [69].
- True IMS residents will have high values for both ratios. Proteins with a low spatial ratio (highly biotinylated by cytosolic APEX) are likely contaminants, even if they show high biotinylation enrichment [69].
This protocol successfully mapped the human mitochondrial IMS proteome with a specificity greater than 94%, identifying 127 proteins including nine novel mitochondrial proteins [69].
Ratiometric Tagging Workflow
The following diagram visualizes the multi-step ratiometric tagging protocol.
The Scientist's Toolkit: Key Reagent Solutions
Successful execution of these quantitative proteomic strategies requires a suite of reliable reagents. The following table details essential materials and their functions.
Table 3: Essential Reagents for Quantitative Proteomics with Proximity Labeling
| Reagent / Material | Function / Application | Key Considerations |
|---|---|---|
| SILAC Media Kits | Provides isotope-labeled essential amino acids (e.g., Lys-8, Arg-10) for metabolic labeling [64] [65]. | Ensure "essential" amino acids to force full incorporation. Verify compatibility with cell line. |
| TMT & iTRAQ Kits | Isobaric chemical tags for multiplexed sample labeling. TMTpro allows 16-plexing [65] [66]. | Beware of ratio compression; use MS3 methods or extensive fractionation to mitigate [66]. |
| APEX2/TurboID Plasmids | Genetically encoded enzymes for proximity labeling. APEX2 for rapid, H₂O₂-activated labeling. TurboID for rapid biotinylation with biotin only [28] [8]. | APEX2 requires optimization of H₂O₂ concentration. TurboID can be toxic due to high biotin depletion [28] [2]. |
| Biotin-Phenol | Substrate for APEX/APEX2 enzymes. Converted to a radical that labels proximal proteins [69] [28]. | Membrane permeability can be a limiting factor in some tissues [28]. |
| Streptavidin Magnetic Beads | High-affinity capture of biotinylated proteins or peptides for purification prior to MS analysis [69] [8]. | Use high-quality beads and stringent wash buffers to minimize non-specific binding. |
| SPS-MS3 Mass Spectrometer | Advanced mass spectrometer configuration that minimizes TMT ratio compression by using a second round of fragmentation [67] [66]. | Reduces the number of quantifiable peptides per unit time but improves quantification accuracy [66]. |
SILAC, TMT, and ratiometric tagging are not universally superior but are uniquely suited to specific biological questions. SILAC excels in cell culture studies where high quantitative precision, especially for post-translational modifications like phosphorylation, is paramount [68]. TMT shines in high-throughput, multi-condition experiments where its multiplexing capacity significantly reduces analytical variability [65] [66]. The innovative ratiometric tagging approach, combining SILAC with proximity labeling, provides a powerful solution for achieving nanometer-scale spatial resolution in mapping subcellular proteomes, overcoming the critical challenge of background labeling [69]. The continued integration of these quantitative strategies with evolving proximity-labeling enzymes will undoubtedly unlock deeper, more precise insights into the spatial and dynamic organization of the proteome.
Proximity labeling (PL) has revolutionized the study of protein-protein interactions, spatial proteomics, and dynamic molecular processes within living systems. This powerful suite of techniques enables researchers to capture intricate spatial and temporal information about cellular environments by covalently tagging neighboring biomolecules with engineered enzymes. As the PL toolbox has expanded, researchers face the critical challenge of selecting the optimal enzyme for their specific experimental needs. This guide provides a comprehensive, data-driven comparison of mainstream PL enzymes, focusing on the four key selection criteria of speed, sensitivity, toxicity, and in vivo applicability to inform researchers, scientists, and drug development professionals in their experimental design.
Technical Comparison of Proximity Labeling Enzymes
Table 1: Key Performance Characteristics of Proximity Labeling Enzymes
| Enzyme | Labeling Time | Catalytic Requirements | Primary Labeling Radius | Toxicity Concerns | In Vivo Compatibility |
|---|---|---|---|---|---|
| BioID | 18-24 hours [18] [2] | ATP | ~10 nm [18] [2] | Low | Excellent (multiple organisms) [47] |
| BioID2 | Several hours [18] [2] | ATP | ~10 nm | Low | Good |
| APEX/APEX2 | 1 minute [18] [2] | H₂O₂, Biotin-phenol [70] | ~20 nm [8] | High (H₂O₂-induced oxidative stress) [18] [2] | Limited (primarily cell culture) [47] |
| TurboID | 10 minutes [18] [2] | ATP, Biotin [47] | ~10 nm | Moderate (biotin depletion at high concentrations) [18] [2] | Excellent (mice, flies, worms, zebrafish, plants) [47] |
| miniTurbo | 10 minutes [47] | ATP, Biotin | ~10 nm | Moderate | Excellent |
| LaccID | 1-2 hours [51] | O₂, Biotin-phenol/BMP [51] | Not specified | Low (uses O₂ instead of H₂O₂) [51] | Promising (tested in fly brain) [51] |
Quantitative Comparison Data
Table 2: Experimental Performance Metrics and Optimization Parameters
| Enzyme | Optimal Substrate Concentration | Temperature Tolerance | Background Labeling | Expression Level Considerations |
|---|---|---|---|---|
| BioID | 50-150 μM biotin | Standard mammalian growth conditions | Low | Tolerates varying expression levels |
| APEX2 | 500 μM biotin-phenol, 1 mM H₂O₂ [70] | Standard conditions | Moderate | Requires sufficient expression for detection [8] |
| TurboID | 50-500 μM biotin [18] [2] | Standard conditions | High (requires optimization) [18] [2] | Functions well at various expression levels |
| LaccID | 250-500 μM BP or BMP [51] | Functions in physiological media [51] | Low to moderate | Selective activity at plasma membrane [51] |
Experimental Protocols for Key Proximity Labeling Enzymes
TurboID/miniTurbo Protocol for Live-Cell Labeling
Step 1: Construct Design and Expression
- Fuse TurboID or miniTurbo to your protein of interest using standard molecular biology techniques [47]
- Transfert into cells and validate proper localization and expression
Step 2: Biotin Administration
- Prepare a 500 μM biotin solution in appropriate cell culture medium
- Add directly to cells and incubate for 10 minutes to several hours depending on experimental needs [18] [2]
- For in vivo applications, administer biotin via appropriate route (injection, feeding) [47]
Step 3: Cell Lysis and Protein Extraction
- Wash cells with PBS to remove excess biotin
- Lyse cells using RIPA buffer or similar formulation with protease inhibitors
Step 4: Streptavidin Affinity Purification
- Incubate lysate with streptavidin-coated magnetic beads
- Wash extensively with lysis buffer to remove non-specifically bound proteins
Step 5: Protein Identification and Analysis
- Perform on-bead tryptic digestion or direct elution for mass spectrometry analysis
- Analyze using LC-MS/MS for protein identification [47]
APEX/APEX2 Labeling Protocol
Step 1: Cell Preparation
- Express APEX/APEX2 fusion construct in target cells
- Pre-incubate with 500 μM biotin-phenol for 30 minutes [70]
Step 2: Hydrogen Peroxide Stimulation
- Add 1 mM H₂O₂ for exactly 1 minute to initiate labeling [70]
- Quench immediately with antioxidant solutions (e.g., ascorbate, Trolox)
Step 3: Protein Capture and Analysis
- Proceed with cell lysis and streptavidin purification as in TurboID protocol
- Analyze by western blot or mass spectrometry
Critical Optimization Parameters
For TurboID, careful optimization of biotin concentration and labeling time is essential to minimize background while maintaining sufficient signal [18] [2]. For APEX2, H₂O₂ concentration must be carefully titrated to balance labeling efficiency against cellular toxicity [70]. Media composition significantly affects LaccID activity, with EBSS providing optimal conditions compared to complete DMEM [51].
Visualization of Proximity Labeling Mechanisms
Enzyme Mechanism Workflow
Experimental Workflow Diagram
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Proximity Labeling Experiments
| Reagent Category | Specific Examples | Function and Application Notes |
|---|---|---|
| PL Enzymes | TurboID, miniTurbo, APEX2, BioID, LaccID [47] [51] | Core labeling enzymes with different performance characteristics; selection depends on experimental priorities |
| Substrates | Biotin (for TurboID/BioID), Biotin-phenol (for APEX), Biotin-methoxyphenol (for LaccID) [70] [51] | Enzyme-specific substrates that become reactive intermediates for protein labeling |
| Cofactors | H₂O₂ (APEX/APEX2), ATP (TurboID/BioID), O₂ (LaccID) [70] [51] | Essential activators for enzymatic reactions; concentration critical for efficiency and toxicity |
| Affinity Matrices | Streptavidin-coated magnetic beads [47] | Critical for purification of biotinylated proteins; ensure high binding capacity and low nonspecific binding |
| Quenching Reagents | Sodium ascorbate, Trolox, Catalase [70] | Essential for stopping peroxidase-based reactions (APEX) to prevent over-labeling and cellular damage |
| Lysis Buffers | RIPA buffer with protease inhibitors | Effective extraction while maintaining protein integrity and interactions |
| Mass Spectrometry | LC-MS/MS systems with high sensitivity | Ultimate detection method for identifying labeled proteins; requires appropriate database search algorithms |
The selection of an appropriate proximity labeling enzyme requires careful consideration of the trade-offs between speed, sensitivity, toxicity, and in vivo applicability. TurboID and miniTurbo currently offer the best balance for most live-cell and in vivo applications, with rapid labeling times and good compatibility across model organisms. APEX2 remains valuable for ultrastructural studies and applications requiring extremely fast labeling, despite its toxicity limitations. The newly developed LaccID presents a promising alternative with reduced toxicity, particularly for cell surface proteomics. Researchers should align their enzyme selection with specific experimental requirements, prioritizing labeling speed for dynamic processes, low toxicity for sensitive biological systems, and proven in vivo performance for whole-organism studies. As the field continues to evolve, further engineering of PL enzymes will likely yield additional tools with enhanced properties and specialized applications.
In the field of intracellular protein research, confidence in experimental results is paramount. Orthogonal validation—the practice of confirming findings using two or more methodologically distinct techniques—provides a powerful framework for achieving this confidence. Among the available tools, Co-Immunoprecipitation (Co-IP) and chemical crosslinking represent complementary methodologies that, when integrated, offer a robust approach for verifying protein-protein interactions (PPIs) and complex formations. This guide objectively compares these techniques and their role in validating emerging proximity-labeling technologies, providing researchers with experimental protocols and data-driven insights to strengthen their scientific conclusions.
Core Principle: Complementary Strengths of Co-IP and Crosslinking
The power of combining Co-IP and crosslinking stems from their fundamental mechanistic differences, which address distinct aspects of protein interaction analysis.
Co-IP functions by using an antibody or affinity tag to capture a protein "bait" and its associated "prey" partners from a cell lysate under mild conditions. This technique is excellent for identifying stable, soluble complexes that maintain their integrity through the purification process [30]. However, it can miss weak or transient interactions that dissociate during cell lysis and washing steps.
Chemical Crosslinking, particularly when coupled with mass spectrometry (XL-MS), introduces covalent bonds between neighboring proteins in living cells before lysis. This "freezes" transient interactions in place, allowing for their subsequent identification even under harsh purification conditions that would normally disrupt them [30]. This makes XL-MS particularly valuable for capturing dynamic complexes.
Table: Fundamental Characteristics of Co-IP and Crosslinking
| Parameter | Co-Immunoprecipitation (Co-IP) | Chemical Crosslinking (XL-MS) |
|---|---|---|
| Interaction Type Captured | Strong, stable complexes | Weak, transient, and stable complexes |
| Spatial Context | Interactions preserved after cell lysis | Interactions in native cellular environment |
| Covalent Modification | No (native complexes) | Yes (crosslinked complexes) |
| Typical Background | Higher (nonspecific binding) | Lower (harsher washing possible) |
| Temporal Resolution | Snapshot after lysis | Snapshot at crosslinking moment |
Direct Comparison: Performance and Capabilities
When evaluated across key performance metrics, Co-IP and crossloading demonstrate distinct operational profiles that inform their optimal application in validation workflows.
Sensitivity and Specificity: Co-IP traditionally suffers from higher background due to nonspecific binding during the enrichment process [30]. Crosslinking enables stronger washing to remove nonspecific binders, thereby improving signal-to-noise ratios [30]. However, crosslinking efficiency varies depending on the reagent chemistry and accessibility of reactive amino acid residues.
Interaction Dynamics: Co-IP is suboptimal for capturing brief interaction events. Crosslinking effectively traps even transient protein-protein interactions through covalent bonding, preserving these associations for analysis [30] [71]. For example, XL-MS has been used to map 6,439 interactions among 2,484 proteins in HEK293 cells, demonstrating its power for large-scale interaction mapping [30].
Complexity of Data Analysis: Co-IP data is relatively straightforward to interpret. Crosslinking data analysis is more challenging due to the increased and more complex search space, though specialized software tools have been developed to address this challenge [30].
Table: Experimental Performance Comparison
| Performance Metric | Co-Immunoprecipitation (Co-IP) | Chemical Crosslinking (XL-MS) |
|---|---|---|
| Transient Interaction Capture | Limited | Excellent |
| Stable Complex Analysis | Excellent | Excellent |
| Background Interference | Moderate to High | Low to Moderate |
| Structural Resolution | Low (proximity within complex) | Medium (residue-level proximity) |
| Multiprotein Complex Mapping | Good | Excellent |
| Typical Experimental Duration | 1-2 days | 2-3 days (including crosslink identification) |
Integration with Proximity-Labeling Enzymes
Proximity-labeling (PL) technologies such as APEX/APEX2 and TurboID have revolutionized the study of subcellular proteomes and interactomes by enabling spatially restricted biotinylation of proximal proteins in living cells [19] [47]. However, as these methods rely on enzyme kinetics and radical diffusion, orthogonal validation remains crucial.
The Validation Pipeline: A typical workflow involves using a PL enzyme (e.g., TurboID fused to a protein of interest) to identify candidate proximal proteins. These candidates are then verified through orthogonal methods:
- Co-IP confirms that the interaction persists under native purification conditions.
- Crosslinking provides evidence that the proteins are in direct physical proximity within the cellular context.
This multi-tiered approach was highlighted in a perspective on integrating PL and crosslinking to study host-virus interactions, where both methods covalently label interacting partners in living cells but through distinct mechanisms, providing complementary evidence [71].
Detailed Experimental Protocols
Co-Immunoprecipitation Protocol
This protocol is adapted from large-scale interaction studies such as the BioPlex project [30].
Key Reagents:
- Lysis Buffer: 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% NP-40, plus protease inhibitors
- Wash Buffer: 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 0.1% NP-40
- Elution Buffer: 0.2 M glycine (pH 2.5) or 2× SDS sample buffer for direct denaturation
Procedure:
- Cell Lysis: Lyse cells in ice-cold lysis buffer (30 minutes with gentle agitation).
- Clarification: Centrifuge at 16,000 × g for 15 minutes at 4°C to remove insoluble material.
- Pre-clearing: Incubate lysate with control beads (e.g., protein A/G) for 30 minutes to reduce nonspecific binding.
- Immunoprecipitation: Incubate pre-cleared lysate with antibody-conjugated beads for 2 hours at 4°C.
- Washing: Wash beads 3-4 times with wash buffer.
- Elution: Elute bound proteins with elution buffer or directly denature in SDS sample buffer.
- Analysis: Analyze by immunoblotting or mass spectrometry.
Chemical Crosslinking Protocol
This protocol is based on methodologies that have successfully mapped thousands of interactions [30].
Key Reagents:
- Crosslinker: DSS (Disuccinimidyl suberate) or equivalent membrane-permeable crosslinker
- Quench Solution: 1 M Tris-HCl (pH 7.5)
- Lysis Buffer: SDS- or urea-based buffer for complete solubilization
Procedure:
- Crosslinking: Treat intact cells with crosslinker (typically 1-5 mM in DMSO) for 30 minutes at room temperature.
- Quenching: Add quench solution to final 50-100 mM Tris and incubate 15 minutes.
- Cell Lysis: Lyse cells in denaturing lysis buffer.
- Enrichment: Enrich bait protein and crosslinked partners by affinity purification under denaturing conditions.
- On-bead Digestion: Digest crosslinked complexes with trypsin directly on beads.
- Mass Spectrometry Analysis: Identify crosslinked peptides using specialized search algorithms such as those implemented in XlinkX or plink [30].
Research Reagent Solutions
Table: Essential Research Reagents for Orthogonal Validation
| Reagent Category | Specific Examples | Function in Experimental Workflow |
|---|---|---|
| Affinity Matrices | Protein A/G Agarose, Streptavidin Beads, Anti-FLAG M2 Agarose | Capture and purify bait protein and associated complexes |
| Crosslinkers | DSS (Disuccinimidyl suberate), DSG (Disuccinimidyl glutarate), formaldehyde | Covalently link proximal proteins in native cellular environment |
| Cell Lysis Reagents | NP-40, RIPA Buffer, Digitonin, SDS | Solubilize proteins while maintaining interactions (varying stringency) |
| Protease Inhibitors | PMSF, Complete Mini EDTA-free Protease Inhibitor Cocktail | Prevent protein degradation during extraction and purification |
| Mass Spectrometry Standards | TMT, SILAC labeled reference peptides | Enable quantitative comparison of protein abundance across samples |
The integration of Co-IP and crosslinking provides a powerful orthogonal validation framework that significantly enhances confidence in protein interaction data, particularly when verifying results from proximity-labeling experiments. While Co-IP confirms interactions under native purification conditions, crosslinking captures transient interactions in living cells, together providing complementary evidence for protein complexes. As proximity-labeling technologies continue to evolve with innovations such as photocatalytic systems like CAT-S [72] and hybridization-proximity methods like HyPro [4], the need for rigorous orthogonal validation only grows more important. By implementing the comparative experimental approaches detailed in this guide, researchers can build robust, verifiable interaction networks that advance our understanding of cellular biology and drug development.
Proximity labeling (PL) has revolutionized the study of biomolecular interactions by enabling the covalent tagging and subsequent identification of proteins and other molecules in living cells. Traditional PL enzymes, primarily biotin ligases like TurboID and peroxidases such as APEX2, have been instrumental in mapping protein-protein interactions, subcellular proteomes, and organelle contact sites [18] [8]. However, these tools possess inherent limitations, including dependence on cytotoxic hydrogen peroxide (for peroxidases), significant background from endogenous biotinylation (for biotin ligases), and sometimes insufficient spatiotemporal precision [73] [19]. To overcome these challenges, the PL toolbox has recently been enriched by several innovative enzymes. This guide objectively compares three emerging classes of PL tools—LaccID, the bacterial tyrosinase BmTyr, and novel light-activated systems—evaluating their performance, experimental parameters, and suitability for specific research applications, particularly in intracellular tagging.
The following table summarizes the key characteristics of these novel enzymes alongside a representative traditional tool for context.
Table 1: Comparison of Emerging and Traditional Proximity Labeling Enzymes
| Enzyme | Class / Activation | Catalytic Requirement | Primary Labeling Site | Key Advantages | Major Limitations |
|---|---|---|---|---|---|
| LaccID [73] [19] | Multicopper Oxidase / O₂ | Oxygen (O₂) | Cell Surface | H₂O₂-free; selective cell surface activity; usable in fixed cells and EM. | Low activity; long labeling time (1-2 hours). |
| BmTyr [19] | Bacterial Tyrosinase / O₂ | Oxygen (O₂) | Intracellular & In Vivo | H₂O₂-free; rapid labeling (≤10 min); low background; good in vivo biocompatibility. | Potential sensitivity to hypoxic conditions. |
| Light-Activated Systems(e.g., LOV-TurboID) [19] | Engineered Biotin Ligase / Light | Blue Light & ATP | Cytoplasm & Nucleus | Excellent spatiotemporal control; reduced background in biotin-rich environments. | Low activity in secretory compartments; requires light delivery. |
| APEX2 [18] [8] | Peroxidase / H₂O₂ | Hydrogen Peroxide (H₂O₂) | Various Intracellular Compartments | Very fast labeling (<1 min); high efficiency; compatible with EM. | H₂O₂ induces oxidative stress/toxicity. |
Detailed Analysis of Emerging Enzymes
LaccID: An Oxygen-Dependent Cell Surface Mapper
LaccID is an engineered multicopper oxidase developed through 11 rounds of directed evolution from an ancestral fungal laccase [73]. Its primary innovation lies in using molecular oxygen instead of toxic hydrogen peroxide to catalyze the one-electron oxidation of aromatic substrates, such as biotin-phenol, generating phenoxyl radicals that covalently tag nearby proteins [73] [74].
A surprising and defining characteristic of LaccID is its selective activity at the plasma membrane in both living and fixed cells, making it particularly suited for mapping cell surface proteomes and interactomes [73] [75]. For instance, researchers have successfully deployed LaccID to map the dynamic surface proteome of T cells engaging with tumor cells via antigen-specific T cell receptors [73] [76]. Furthermore, LaccID serves as a genetically encodable tag for electron microscopy, enabling visualization of cell surface features in mammalian cell culture and fly brain tissue [73].
However, a significant drawback is its relatively low enzymatic activity. While APEX2 or TurboID can label in minutes, LaccID requires 1 to 2 hours of incubation with substrate to yield detectable signals, limiting its ability to capture rapid dynamic processes [19]. Its labeling efficiency may also be compromised under hypoxic conditions due to its dependence on oxygen [19].
Table 2: Key Experimental Parameters for LaccID Proximity Labeling
| Parameter | Recommended Condition |
|---|---|
| Labeling Time | 1 - 2 hours [19] |
| Optimal Buffer | Earle's Balanced Salt Solution (EBSS); activity is impaired by thiols in complete media [73] |
| Key Substrate | Biotin-phenol (Biotin-methoxyphenol showed higher activity) [73] |
| Cell Status | Works in both living and fixed cells [73] |
BmTyr: A Rapid and Biocompatible Tyrosinase
The bacterial tyrosinase from Bacillus megaterium (BmTyr) represents another H₂O₂-free alternative that utilizes molecular oxygen for catalysis [19]. It enables rapid (≤10 minutes) and low-background protein labeling, addressing the speed limitations of LaccID [19] [8].
A key advantage of BmTyr is its demonstrated biocompatibility for in vivo applications. Studies have shown its effectiveness in labeling plasma proteins in vivo and profiling region-specific proteomes within the mouse brain, with improved performance over existing PL enzymes [19]. This robust activity in live animals highlights its potential for physiological studies. Furthermore, BmTyr has been adapted for ligand-directed PL, allowing researchers to map the proximal proteomes of endogenous neurotransmitter receptors in the live mouse brain without the need for genetic fusion [19].
Like LaccID, its oxygen-dependent mechanism means labeling efficiency could be reduced in hypoxic tissues or cellular microenvironments [19].
Light-Activated Systems: Precision through Optogenetics
Light-activated PL systems provide unparalleled spatiotemporal control over the labeling process. A prime example is LOV-TurboID, which incorporates a light-sensitive LOV domain into the TurboID enzyme [19]. This design keeps the enzyme inactive in the dark and triggers biotinylation only upon exposure to low-intensity blue light.
This optogenetic control offers two major benefits. First, it drastically reduces background labeling in biotin-rich environments, such as neurons, which is a common challenge with conventional TurboID [19]. Second, it allows researchers to capture molecular interactions within precise time windows, enabling the study of highly dynamic cellular events. However, this system is not without its weaknesses; it shows barely any activity within secretory compartments and has relatively low targeting efficiency when directed to the mitochondrial matrix [19].
Another innovative light-dependent system is the cascade reaction-based PL developed by Pan et al. This method uses a singlet oxygen photosensitizing protein under blue light to convert ambient oxygen into H₂O₂, which in turn activates APEX2 [19]. This creates a self-contained, light-triggered system that does not require the addition of exogenous H₂O₂, achieving high spatiotemporal resolution with a labeling time of less than 10 seconds [19].
Experimental Protocols for Novel Enzymes
Protocol: Cell Surface Proteome Mapping with LaccID
This protocol outlines the key steps for using LaccID to map the surface proteome of live cells, such as T cells [73].
- Construct Design and Expression: Fuse the LaccID gene to a secretion signal sequence and a transmembrane anchor (e.g., from CD4) for robust cell surface expression. Transfect the construct into your target cells (e.g., HEK293T cells or T cells).
- Validation of Expression: Confirm successful expression and surface localization of the LaccID fusion protein via anti-tag (e.g., V5) western blotting and/or immunofluorescence.
- Labeling Reaction:
- Wash the cells expressing LaccID with a pre-warmed physiological buffer like Earle's Balanced Salt Solution (EBSS). Note that complete culture media (e.g., DMEM, RPMI) containing thiols can inhibit LaccID activity.
- Incubate the cells with the Biotin-Phenol (BP) or Biotin-Methoxyphenol (BMP) substrate dissolved in EBSS for 1-2 hours at 37°C [73].
- Reaction Termination and Cell Lysis: Remove the labeling solution and wash the cells thoroughly with cold buffer containing quenching agents (e.g., ascorbate and Trolox) to stop the reaction. Lyse the cells using RIPA buffer supplemented with protease inhibitors.
- Streptavidin Affinity Purification: Incubate the cell lysate with streptavidin-coated magnetic beads to capture the biotinylated proteins.
- On-Bead Digestion and Mass Spectrometry Analysis: Wash the beads stringently, digest the captured proteins with trypsin, and analyze the resulting peptides by liquid chromatography-tandem mass spectrometry (LC-MS/MS) for protein identification and quantification.
Protocol: Optogenetic Control with LOV-TurboID
This protocol describes the use of LOV-TurboID for spatially or temporally controlled proximity labeling in living cells [19].
- Targeting and Expression: Fuse the LOV-TurboID construct to your protein of interest or a specific organelle targeting signal. Express the construct in the desired cell line.
- Substrate Incubation: Add biotin to the culture medium to ensure substrate availability. The concentration and incubation time should be optimized to minimize background in the dark state.
- Blue Light Activation: Expose the cells to low-intensity blue light to induce a conformational change in the LOV domain, activating TurboID. The duration of light exposure (e.g., seconds to minutes) defines the temporal window for labeling.
- Sample Processing: Following light induction, wash the cells, harvest, and lyse them. The subsequent steps for streptavidin enrichment and MS analysis are identical to those in the LaccID protocol (steps 5-6).
Visualization of Workflows and Mechanisms
The following diagrams illustrate the fundamental mechanisms and experimental workflows for the highlighted enzymes.
Mechanism of LaccID and BmTyr
Diagram 1: O₂-Dependent Enzyme Mechanism
LOV-TurboID Activation Workflow
Diagram 2: Light-Activated Proximity Labeling
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Proximity Labeling Experiments
| Reagent / Solution | Function in Experiment |
|---|---|
| LaccID, BmTyr, or LOV-TurboID Plasmid | Genetically encodable enzyme for catalyzing the proximity labeling reaction [73] [19]. |
| Biotin-Phenol (BP) | Aromatic substrate that is oxidized to form a reactive radical, which covalently tags proximal proteins with biotin [73] [19]. |
| Earle's Balanced Salt Solution (EBSS) | A thiol-free physiological buffer used for LaccID labeling to prevent inhibition of enzyme activity [73]. |
| Streptavidin-Magnetic Beads | Solid-phase affinity matrix for the highly specific capture and purification of biotinylated proteins from complex cell lysates [18] [8]. |
| Sodium Ascorbate / Trolox | Radical quenchers added to washing buffers to terminate the labeling reaction and reduce background from diffused radicals [73]. |
| DIG-Modified Antisense Oligonucleotides | For hybridization-proximity labeling (HyPro); used to recruit the labeling enzyme to specific RNA molecules [4]. |
| Hydrogen Peroxide (H₂O₂) | Essential co-substrate for traditional peroxidase-based PL enzymes like APEX2 (but not for LaccID or BmTyr) [18] [19]. |
The development of LaccID, BmTyr, and light-activated systems like LOV-TurboID marks a significant step forward in the PL field, offering researchers a more diverse and sophisticated toolkit. The choice of enzyme now depends on a clear set of experimental priorities: LaccID is the specialist for H₂O₂-free cell surface mapping and EM, BmTyr excels in rapid, low-background in vivo proteomics, and light-activated systems provide supreme spatiotemporal precision for dynamic studies.
Future innovations will likely focus on further improving the activity and specificity of these novel enzymes, developing systems responsive to other endogenous signals (e.g., other ions or metabolites), and creating even more compact tags to minimize functional disruption of the target protein [19]. As these tools continue to evolve, they will undoubtedly unlock deeper insights into the intricate molecular organization of living systems.
Conclusion
The strategic selection of a proximity labeling enzyme is paramount for successful intracellular tagging, with the choice hinging on specific experimental goals regarding temporal resolution, spatial precision, and biological context. Foundational peroxidase tools like APEX2 offer unmatched speed for capturing transient interactions, while evolved biotin ligases like TurboID provide superior sensitivity for in vivo applications. As the field advances, the emergence of environment-responsive, light-activated, and endogenous-targeting enzymes is pushing the boundaries of spatial proteomics. These innovations promise to unlock deeper insights into dynamic cellular processes, accelerate drug target discovery, and ultimately bridge our understanding from molecular interactions to physiological function and dysfunction in human disease. Future developments will likely focus on further reducing background, improving spatiotemporal control, and expanding applications in clinical samples.
References
- 1. Filling the void: Proximity-based labeling of proteins in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5077660/]
- 2. Bridging molecular and cellular neuroscience with ... [https://www.nature.com/articles/s12276-025-01491-4]
- 3. Efficient proximity labeling in living cells and organisms ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6126969/]
- 4. Enhanced hybridization-proximity labeling discovers ... [https://www.nature.com/articles/s41467-025-64282-5]
- 5. A proximity-labeling-based approach to directly detect ... [https://www.sciencedirect.com/science/article/pii/S2162253125001568]
- 6. Rapid, biochemical tagging of cellular activity history in vivo [https://www.nature.com/articles/s41592-024-02375-7]
- 7. Next-Generation Protein–Ligand Interaction Networks: APEX ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12286260/]
- 8. The development of proximity labeling technology and its ... [https://biosignaling.biomedcentral.com/articles/10.1186/s12964-023-01310-1]
- 9. Cell-type and subcellular compartment-specific APEX2 ... [https://www.nature.com/articles/s41467-021-25144-y]
- 10. HRP vs. APEX2 - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10828542/]
- 11. An enzymatic cascade enables sensitive and specific ... [https://www.nature.com/articles/s41467-025-65405-8]
- 12. Comparative Application of BioID and TurboID for Protein- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7290721/]
- 13. Comparative Application of BioID and TurboID for Protein ... [https://www.mdpi.com/2073-4409/9/5/1070]
- 14. TurboID functions as an efficient biotin ligase for BioID ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9643680/]
- 15. TurboID-Based Proximity Labeling: A Method to Decipher ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12363416/]
- 16. Control of TurboID-dependent biotinylation intensity in ... [https://www.sciencedirect.com/science/article/pii/S1874391923000751]
- 17. Employing Expression-Matched Controls Enables High- ... [https://www.sciencedirect.com/science/article/pii/S1535947625001008]
- 18. Bridging molecular and cellular neuroscience with proximity ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12322126/]
- 19. Evolving advances of proximity labeling in capturing ... [https://www.sciencedirect.com/science/article/abs/pii/S1367593125000614]
- 20. Directed Evolution: Past, Present and Future - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4344831/]
- 21. -dependent Proximity methods for proteomic profiling in living... labeling [https://pmc.ncbi.nlm.nih.gov/articles/PMC8142282/]
- 22. A bacterial display system for effective selection of protein ... [https://www.nature.com/articles/s41598-019-40984-x]
- 23. Directed evolution [https://en.wikipedia.org/wiki/Directed_evolution]
- 24. A primer to directed evolution: current methodologies and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10074555/]
- 25. Directed evolution of the substrate specificity of biotin ligase [https://pubmed.ncbi.nlm.nih.gov/24375025/]
- 26. Rapid, biochemical tagging of cellular activity history... | Nature Methods [https://www.nature.com/articles/s41592-024-02375-7?error=cookies_not_supported&code=1148c1ea-1917-4c97-a308-1ad1fdd8b7bc]
- 27. Super-resolution proximity labeling with enhanced direct ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11082246/]
- 28. Advances in enzyme-mediated proximity labeling and its ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8825456/]
- 29. Proximity Labeling: A Powerful Tool for Protein Complex ... [https://blog.addgene.org/proximity-labeling-a-powerful-tool-for-protein-complex-purification-and-proteomic-mapping]
- 30. Proximity labeling for investigating protein- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10782847/]
- 31. The development of proximity labeling technology and its ... [https://pubmed.ncbi.nlm.nih.gov/37777761/]
- 32. Proximity Labeling in Plants - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10576617/]
- 33. Super-resolution proximity labeling with enhanced direct ... [https://www.nature.com/articles/s42003-024-06112-w]
- 34. Proximity labeling uncovers the synaptic proteome under ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12325354/]
- 35. Endogenous TOM20 Proximity Labeling: A Swiss-Knife for ... [https://www.mdpi.com/1422-0067/24/11/9604]
- 36. Deciphering Spatial Protein–Protein Interactions in Brain ... [https://www.sciencedirect.com/science/article/pii/S1535947622002304]
- 37. A proximity labeling strategy enables proteomic analysis of ... [https://www.sciencedirect.com/science/article/pii/S2589004223012361]
- 38. Multidimensional tracking of GPCR signaling via ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5514552/]
- 39. A Network of G Protein αi Signaling Partners is Revealed by ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9082328/]
- 40. Spatiotemporally resolved GPCR interactome uncovers ... [https://www.sciencedirect.com/science/article/pii/S2451945625001291]
- 41. Interrogating Kinase–Substrate Relationships with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9142857/]
- 42. Identification of Endogenous Kinase Substrates by ... [https://www.sciencedirect.com/science/article/pii/S1535947621000918]
- 43. Improving split‐HaloTag through computational protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12006747/]
- 44. Molecular Mechanisms of Intracellular Delivery ... [https://link.springer.com/article/10.1007/s40820-023-01313-0]
- 45. Enzyme-mediated proximity labeling reveals the co ... [https://pubs.rsc.org/en/content/articlehtml/2025/cb/d4cb00155a]
- 46. An integrated transcriptomic and proteomic map of the ... [https://www.nature.com/articles/s41467-025-63119-5]
- 47. From Vision to Reality: A Perspective on the Innovative and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12446214/]
- 48. Characterization of the intracellular neurexin interactome by in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10802952/]
- 49. Development and Comparative Evaluation of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8895409/]
- 50. Proximity labeling and affinity capture compared - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9791439/]
- 51. Directed evolution of LaccID for cell surface proximity ... [https://www.nature.com/articles/s41589-025-01973-6]
- 52. High throughput generation of tagged stable cell lines for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4785821/]
- 53. A Simple and Scalable Strategy for Analysis of ... [https://www.nature.com/articles/s41598-020-65832-1]
- 54. Endogenous tagging using split mNeonGreen in human ... [https://elifesciences.org/articles/92819]
- 55. Epitope tagging of endogenous genes in diverse human ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2577350/]
- 56. The development of proximity labeling technology and its applications in mammals, plants, and microorganisms | Cell Communication and Signaling [https://link.springer.com/article/10.1186/s12964-023-01310-1]
- 57. Proximity labeling - Wikipedia [https://en.wikipedia.org/wiki/Proximity_labeling]
- 58. In vivo interactome profiling by enzyme‐catalyzed proximity labeling | Cell & Bioscience | Full Text [https://cellandbioscience.biomedcentral.com/articles/10.1186/s13578-021-00542-3]
- 59. A Tutorial Review of Labeling Methods in Mass ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11342459/]
- 60. Proximity labeling uncovers the synaptic proteome under ... [https://www.frontiersin.org/journals/cellular-neuroscience/articles/10.3389/fncel.2025.1638627/full]
- 61. Comprehensive dataset of interactors for the entire PARP ... [https://www.nature.com/articles/s41597-025-04722-5]
- 62. Report APEX2 proximity labeling of RNA in bacteria [https://www.sciencedirect.com/science/article/pii/S2667237525002425]
- 63. Intracristal space proteome mapping using super- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12368266/]
- 64. Stable isotope labeling by amino acids in cell culture - Wikipedia [https://en.wikipedia.org/wiki/Stable_isotope_labeling_by_amino_acids_in_cell_culture]
- 65. iTRAQ, Comparing and TMT | Silantes SILAC [https://silantes.com/itraq-tmt-silac/]
- 66. The Rockefeller University » MS-based Relative Quantitation [https://www.rockefeller.edu/proteomics/proteomics/ms-based-quantitation/]
- 67. Journal Club: Time-Resolved Analysis of Proteome Dynamics by... [https://www.thermofisher.com/us/en/home/references/newsletters-and-journals/bioprobes-journal-of-cell-biology-applications/bioprobes-76/bioprobes-76-journal-club-time-resolved-proteome-analysis-tmt-silac-hyperplexing.html]
- 68. Systematic Comparison of Label-Free, SILAC , and TMT Techniques... [https://pubmed.ncbi.nlm.nih.gov/31814417/]
- 69. mapping of the human mitochondrial intermembrane space... Proteomic [https://pmc.ncbi.nlm.nih.gov/articles/PMC4743503/]
- 70. Near-infrared Molecular Probes for In Vivo Imaging - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3334312/]
- 71. An integrated approach using proximity labelling and chemical crosslinking to probe in situ host-virus protein–protein interactions - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11649372/]
- 72. Bioorthogonal photocatalytic proximity labeling in primary ... [https://www.nature.com/articles/s41467-024-46985-3]
- 73. Directed evolution of LaccID for cell surface proximity labeling ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12643941/]
- 74. Directed evolution of LaccID for cell surface proximity labeling ... [https://pubmed.ncbi.nlm.nih.gov/40751001/]
- 75. [PDF] Directed evolution of LaccID for cell surface proximity ... [https://www.semanticscholar.org/paper/Directed-evolution-of-LaccID-for-cell-surface-and-Lee-Roh/3ca7bbae9310a70e9f0c0ae87e50f093f37859b1]
- 76. Directed evolution of the multicopper oxidase laccase for cell ... [https://pubmed.ncbi.nlm.nih.gov/39554088/]