Overcoming Cell Permeability Barriers: Advanced Strategies for Successful Intracellular Target Assays
This article provides a comprehensive guide for researchers and drug development professionals facing the critical challenge of cell permeability in intracellular target assays.
Overcoming Cell Permeability Barriers: Advanced Strategies for Successful Intracellular Target Assays
Abstract
This article provides a comprehensive guide for researchers and drug development professionals facing the critical challenge of cell permeability in intracellular target assays. It explores the fundamental biological barriers that hinder compound entry, surveys traditional and cutting-edge methodological approaches from Caco-2 models to organ-on-a-chip systems and AI-driven prediction tools. The content details practical strategies for troubleshooting and optimizing permeability, including structural modification and advanced delivery systems, while also addressing validation techniques and comparative analysis of assay data. By synthesizing foundational knowledge with advanced applications, this resource aims to equip scientists with the multidisciplinary strategies needed to accurately profile compound activity against intracellular targets and accelerate therapeutic development.
Understanding the Cellular Gatekeeper: Fundamental Barriers to Intracellular Compound Delivery
Troubleshooting Guide: Common Issues in Permeability and Engagement Assays
| Problem Area | Specific Issue | Possible Causes | Recommended Solutions |
|---|---|---|---|
| Cellular Permeability | Compound shows high biochemical potency but low cellular potency (cell drop-off) [1]. | Low membrane permeability; efflux by transporters like P-glycoprotein; extensive nonspecific cellular binding [1]. | Measure intracellular bioavailability (Fic); use efflux transporter inhibitors (e.g., cyclosporine A) in assays; optimize logP and MW to improve passive diffusion [1]. |
| Thermal Shift Assays (TSA) | Irregular melt curves in DSF [2]. | Compound intrinsic fluorescence; compound-dye interactions; poor compound solubility; incompatible buffer components (e.g., detergents) [2]. | Use a concentration-matched compound control well; test different fluorescent dyes; optimize buffer conditions and DMSO concentration; check compound solubility [2]. |
| Cellular Thermal Shift Assay (CETSA) | No shift observed in whole-cell CETSA, but shift is seen in cell lysate [2]. | Inefficient cell membrane permeability of the test compound [2]. | Confirm compound permeability using a separate method (e.g., Fic); extend compound incubation time with cells; use a lysate CETSA as a positive control [2]. |
| General TSA Performance | Poor protein stabilization or low signal-to-noise ratio [2]. | Protein instability in assay buffer; inappropriate protein concentration; test compound affects assay pH or ionic strength [2]. | Screen different protein buffers for optimal stability; ensure protein is soluble and not aggregated; include a stabilizing positive control ligand [2]. |
| Data Interpretation | Inconsistent Tm values or poor curve fits [2]. | Protein aggregation at starting temperature; low signal intensity [2]. | Visually confirm sample clarity before runs; optimize protein and dye concentrations; use data analysis software that allows for manual baseline adjustment [2]. |
Frequently Asked Questions (FAQs)
Q1: What is "intracellular bioavailability (Fic)" and why is it a better metric than artificial membrane permeability (PAMPA) for predicting cellular activity?
A: Intracellular bioavailability (Fic) represents the fraction of the extracellularly added drug that is bioavailable inside the cell in its unbound form, available to engage the target [1]. It is a direct measure that incorporates the net effect of membrane permeability, active transport, metabolism, and nonspecific binding [1]. While artificial membrane permeability (e.g., from PAMPA) only measures passive diffusion rates, Fic has been shown to correlate much better with actual cellular potency, explaining why some compounds with good PAMPA data still fail in cellular assays [1].
Q2: In a CETSA experiment, my compound works in cell lysate but not in intact cells. What does this mean, and what should I do next?
A: This is a classic indication that your compound has poor cell membrane permeability [2]. The lysate experiment confirms that the compound can bind and stabilize the target protein when the membrane barrier is removed. Your next steps should be to:
- Confirm Permeability: Use a direct method to measure the compound's intracellular concentration or bioavailability (Fic) [1].
- Modify the Compound: Investigate structural modifications to improve permeability, such as reducing logP, molecular weight, or the number of hydrogen bond donors/acceptors [3].
- Check for Efflux: Perform the cellular assay in the presence of an efflux transporter inhibitor (e.g., cyclosporine A) to see if potency improves [1].
Q3: What are the main benefits and limitations of using Thermal Shift Assays (TSAs) like DSF and CETSA for measuring target engagement?
A:
- Benefits: TSAs are label-free, meaning neither the drug nor the protein needs to be modified, which preserves native interactions [2]. They can be adapted to high-throughput formats (especially DSF) and can be used in both biochemical (DSF) and more physiologically relevant cellular (CETSA) settings [2].
- Limitations: The elevated temperatures used can affect the binding kinetics of some compounds and are non-physiological [2]. For CETSA, the readout is protein aggregation, which is an indirect measure of binding and may not be suitable for all proteins [2]. The technique also requires a specific and sensitive method for detecting the protein of interest, such as a good antibody for Western blotting [2].
Experimental Protocols: Key Methodologies
Protocol 1: Measuring Intracellular Bioavailability (Fic)
This protocol is used to quantitatively determine the fraction of unbound drug inside the cell [1].
- Cell Preparation: Seed the relevant cell type (e.g., PBMCs, HeLa) in a standard culture plate and grow to the desired confluence.
- Compound Incubation: Expose the cells to the test compound at a relevant concentration for a specified time period.
- Separation and Lysis: After incubation, rapidly separate the cells from the medium by centrifugation. Wash the cell pellet with a cold buffer to remove compound adhering to the outer membrane. Lyse the cells.
- Quantification: Measure the total intracellular drug concentration using a sensitive method like LC-MS/MS.
- Determine Unbound Fraction: Use a technique like equilibrium dialysis or ultrafiltration on the cell lysate to determine the fraction of unbound drug (fu,cell) and the cellular compound accumulation (Kp).
- Calculation: The intracellular bioavailability (Fic) is calculated from these parameters. A high Fic indicates good intracellular target access [1].
Protocol 2: Differential Scanning Fluorimetry (DSF) for Initial Compound Screening
This is a high-throughput method to identify binders to a purified recombinant protein [2].
- Sample Preparation:
- Prepare a master mix containing purified recombinant protein and a fluorescent dye (e.g., SYPRO Orange) in an optimized buffer. Detergents should be avoided as they can interfere with the dye [2].
- Dispense the master mix into a multi-well plate.
- Add test compounds and a positive control (known binder) to respective wells. Include a DMSO-only well as a negative control.
- Run the Assay: Place the plate in a real-time PCR instrument or a dedicated thermal cycler. Program a thermal ramp (e.g., from 25°C to 95°C with a gradual increase of 0.5-1°C per minute) while monitoring fluorescence.
- Data Analysis: Plot fluorescence versus temperature to generate melt curves for each well. Calculate the melting temperature (Tm) for each condition. A significant shift in Tm (ΔTm) in the presence of a compound indicates binding and stabilization of the protein structure [2].
Conceptual and Workflow Diagrams
Intracellular Drug Engagement Pathway
Troubleshooting Low Cellular Potency
The Scientist's Toolkit: Essential Research Reagents
| Reagent / Material | Function in Assays | Key Considerations |
|---|---|---|
| SYPRO Orange Dye | A polarity-sensitive fluorescent dye used in DSF to bind hydrophobic patches of unfolded proteins [2]. | Incompatible with detergents; can interact with some test compounds, causing artifacts [2]. |
| Caco-2 Cell Line | A human colon carcinoma cell line that forms polarized monolayers, used as a standard in vitro model for predicting intestinal permeability [3]. | Requires long culture time (21 days) to fully differentiate; results can be variable between labs [3]. |
| Cyclosporine A | A pan-inhibitor of active efflux transporters (e.g., P-gp) [1]. | Used in mechanistic studies to confirm if poor permeability/Fic is due to active efflux [1]. |
| Heat-Stable Proteins (SOD1, APP-αCTF) | Used as loading controls in Western blot-based TSAs (PTSA, CETSA) for data normalization [2]. | Must remain soluble at high temperatures while the target protein melts/aggregates [2]. |
| Nanoluciferase (NanoLuc) | A small, bright luciferase used in NanoBRET assays to tag target proteins for studying intracellular binding kinetics and target engagement in live cells [4]. | Provides a highly sensitive, bioluminescent readout for real-time kinetic measurements [4]. |
Technical Support Center: Troubleshooting Intracellular Assay Permeability
FAQ & Troubleshooting Guide
Q1: My test compound shows high in vitro potency but no cellular activity. Are cellular defense mechanisms preventing its entry? A: This is a classic symptom of poor cell permeability. To diagnose, follow this workflow:
- Step 1: Rule out Efflux. Treat cells with an efflux transporter inhibitor (e.g., Verapamil for P-gp, Ko143 for BCRP) and re-run your cellular assay. A significant increase in activity suggests active efflux is the primary defense mechanism.
- Step 2: Assess Passive Permeability. Perform a parallel artificial membrane permeability assay (PAMPA). Compare the permeability (Pe) of your compound to known standards (see Table 1).
- Step 3: Determine Route of Entry. Use inhibitors or modulators of specific pathways (see Table 2) to pinpoint the transport mechanism.
Q2: How can I distinguish between paracellular and transcellular passive diffusion? A: The primary differentiator is molecular weight (MW) and the presence of tight junctions.
- Paracellular Transport: This pathway is generally restricted to small, hydrophilic compounds (typically < 500 Da). It is highly dependent on the cell type and the integrity of its tight junctions.
- Transcellular Passive Diffusion: This is the preferred route for lipophilic, uncharged compounds. It is not size-restricted in the same way, but larger molecules diffuse more slowly.
- Experimental Protocol: Use a high-integrity cell monolayer (e.g., Caco-2 or MDCK). Measure the apparent permeability (Papp) and the transepithelial electrical resistance (TEER). A high Papp for a small, polar molecule coupled with a low TEER value indicates significant paracellular leakage. A compound with high Papp that does not affect TEER is likely using the transcellular route.
Q3: My compound is a substrate for an uptake transporter. How can I leverage this for intracellular delivery? A: You can design a "pro-drug" strategy. Chemically modify your compound to make it a better substrate for a specific, highly expressed uptake transporter (e.g., peptide or nucleotide transporters). The pro-drug is actively transported into the cell, where endogenous enzymes cleave the modification to release the active parent compound.
Q4: What controls are essential for a reliable carrier-mediated transport assay? A: Always include these controls:
- Negative Control: A compound known not to use the transporter of interest.
- Positive Control: A known high-affinity substrate for the transporter.
- Inhibitor Control: Assay in the presence of a specific chemical inhibitor to confirm transporter dependence.
- Temperature Control: Perform the assay at 4°C (to inhibit active transport) and 37°C. A significant reduction in uptake at 4°C confirms an energy-dependent process.
Table 1: Benchmarking Permeability in Standard Assays
| Assay Type | Measures | Typical Output | High Permeability Benchmark | Interpretation |
|---|---|---|---|---|
| PAMPA | Passive Transcellular | Effective Permeability (Pe) | Pe > 1.5 x 10⁻⁶ cm/s | Predicts passive diffusion potential. |
| Caco-2 | Combined Routes | Apparent Permeability (Papp) | Papp (A-B) > 10 x 10⁻⁶ cm/s | Models intestinal absorption; includes efflux. |
| P-gp Assay | Active Efflux | Efflux Ratio (B-A/A-B) | Efflux Ratio < 2.5 | Low risk of being a P-gp substrate. |
Table 2: Key Transporter Inhibitors for Pathway Diagnosis
| Transport Pathway | Representative Inhibitor | Common Working Concentration | Primary Use in Troubleshooting |
|---|---|---|---|
| P-glycoprotein (P-gp) | Verapamil | 50 - 100 µM | To confirm/reverse P-gp mediated efflux. |
| BCRP | Ko143 | 1 - 5 µM | To confirm/reverse BCRP mediated efflux. |
| OATPs | Rifampicin | 10 - 50 µM | To inhibit OATP-mediated uptake. |
| OCTs | Cimetidine | 100 - 500 µM | To inhibit OCT-mediated uptake. |
Experimental Protocols
Protocol 1: Caco-2 Permeability Assay to Diagnose Transport Routes
Objective: To determine the apparent permeability (Papp) of a test compound and identify the contribution of transcellular, paracellular, and active transport pathways.
Materials:
- Caco-2 cell monolayers (21-25 days post-seeding, TEER > 300 Ω·cm²)
- Transport buffer (HBSS or DPBS, pH 7.4)
- Test compound
- Reference compounds (e.g., Propranolol for high permeability, Atenolol for low permeability)
- Transwell plates (e.g., 12-well, 1.12 cm² insert area)
- LC-MS/MS system for quantification
Method:
- Pre-incubation: Equilibrate Caco-2 monolayers and transport buffer at 37°C for 20 min.
- Dosing: Add the test compound (typically 5-10 µM) to the donor compartment (Apical for A-B, Basolateral for B-A).
- Inhibition (Optional): Co-incubate with a transporter inhibitor (see Table 2) in both donor and receiver compartments.
- Incubation: Place the plate on an orbital shaker (50-60 rpm) at 37°C for a set time (e.g., 60-120 min).
- Sampling: At the end time point, collect samples from both donor and receiver compartments.
- Analysis: Quantify compound concentration using LC-MS/MS.
- Calculation: Calculate Papp (cm/s) using the formula: Papp = (dQ/dt) / (A * C₀), where dQ/dt is the transport rate, A is the membrane area, and C₀ is the initial donor concentration.
- Interpretation: Calculate the Efflux Ratio (Papp B-A / Papp A-B). An ER > 2.5 suggests active efflux.
Protocol 2: PAMPA for Assessing Passive Transcellular Permeability
Objective: To measure the intrinsic passive transcellular permeability of a compound, independent of active transporters or paracellular pathways.
Materials:
- PAMPA plate system (e.g., a donor plate and an acceptor plate separated by a membrane)
- Phospholipid solution (e.g., in dodecane)
- Test and reference compounds
- PBS buffer (pH 7.4)
- UV plate reader or LC-MS/MS
Method:
- Membrane Formation: Impregnate the filter membrane with the phospholipid solution to create an artificial lipid bilayer.
- Dosing: Add the test compound solution to the donor well.
- Assembling: Carefully place the acceptor plate on top, ensuring no air bubbles.
- Incubation: Incubate the assembled plate for 2-6 hours at room temperature or 37°C.
- Sampling: Separate the plates and sample from both donor and acceptor wells.
- Analysis: Quantify the compound in both compartments.
- Calculation: Determine the effective permeability (Pe) using a specific model provided by the PAMPA kit manufacturer or standard equations.
Visualization of Pathways and Workflows
Diagram 1: Cellular Transport Pathways
Diagram 2: Permeability Troubleshooting Workflow
The Scientist's Toolkit
| Research Reagent / Material | Function in Permeability Research |
|---|---|
| Caco-2 Cells | A human colon adenocarcinoma cell line that spontaneously differentiates to form tight junctions and express key transporters; a gold-standard model for intestinal permeability and efflux. |
| MDCK-II Cells | Madin-Darby Canine Kidney cells; form tighter monolayers faster than Caco-2. Often transfected with human transporters (e.g., MDCK-MDR1) for specific efflux studies. |
| Verapamil | A calcium channel blocker and well-established, non-specific chemical inhibitor of the P-glycoprotein (P-gp) efflux transporter. |
| Ko143 | A potent and specific chemical inhibitor of the Breast Cancer Resistance Protein (BCRP/ABCG2) efflux transporter. |
| PAMPA Plate | A high-throughput screening tool using an artificial phospholipid membrane to measure intrinsic passive transcellular permeability, free from cellular complications. |
| LC-MS/MS | Liquid Chromatography with Tandem Mass Spectrometry; the gold-standard analytical method for sensitive and specific quantification of test compounds in complex biological matrices from permeability assays. |
FAQs on Molecular Properties and Permeability
Q1: What are the key molecular properties that govern a compound's permeability? The primary molecular properties governing permeability are lipophilicity (often measured as LogP or LogD), molecular size (often represented by Molecular Weight or Polar Surface Area), and hydrogen-bonding capacity (count of Hydrogen Bond Donors and Acceptors). Permeability is optimized by a balance of these properties, such as moderate lipophilicity and limited polar surface area, which facilitate passive diffusion across cell membranes [5] [6].
Q2: Why does a compound with high biochemical potency sometimes show weak activity in cellular assays? This discrepancy, often termed "cell drop off," is frequently due to low intracellular bioavailability (Fic). A compound may bind perfectly to an isolated target, but if it cannot efficiently cross the cell membrane to reach that target inside the cell, its cellular potency will be low. This can be caused by poor passive permeability, efflux by transporter proteins, or extensive intracellular binding [1].
Q3: How can intramolecular hydrogen bonds (IMHBs) improve a compound's permeability? Intramolecular hydrogen bonds can effectively "shield" polar groups (H-bond donors and acceptors) from the aqueous environment, reducing the molecule's overall apparent polarity. This can enhance lipophilicity and membrane permeability, a strategy sometimes referred to as "polarity shielding" [7] [8]. This is particularly valuable for compounds beyond the Rule of 5 (bRo5) [6].
Q4: Is the Rule of 5 (Ro5) still relevant for designing permeable compounds? Yes, the Ro5 provides a valuable guideline for compounds intended for oral administration. However, permeable and orally bioavailable drugs can be discovered far beyond Ro5 space (bRo5), especially for difficult targets. Achieving this often requires deliberate design strategies, such as the formation of intramolecular hydrogen bonds and managing conformational flexibility [6].
Q5: How does measured intracellular bioavailability (Fic) differ from artificial membrane permeability (e.g., PAMPA)? Fic is a cell-based measurement that quantifies the actual unbound drug concentration inside the cell. It is a net result of membrane permeability, carrier-mediated transport, metabolism, and nonspecific cellular binding. In contrast, PAMPA measures passive diffusion across an artificial membrane and does not account for complex biological processes. Studies show a poor correlation between PAMPA permeability and Fic, underscoring the value of direct intracellular measurement for predicting target engagement [1].
Troubleshooting Guides
Issue 1: Poor Correlation Between Biochemical and Cellular Potency
Potential Causes and Solutions:
Cause: Low Intracellular Bioavailability. The compound fails to accumulate inside the cell in its unbound, active form.
- Solution: Measure the intracellular bioavailability (Fic) directly. A label-free method using mass spectrometry can determine the unbound intracellular concentration, providing a direct metric for target access [1] [9].
- Solution: Investigate the impact of efflux transporters. Conduct permeability assays (e.g., Caco-2) in the presence and absence of a pan-inhibitor like cyclosporine A. A significant increase in permeability in the presence of the inhibitor suggests active efflux is limiting cellular exposure [1].
Cause: Suboptimal Physicochemical Properties.
Table 1: Molecular Property Guidelines for Permeability
| Molecular Property | Traditional (Ro5) Space | Beyond Rule of 5 (bRo5) Space | Impact on Permeability |
|---|---|---|---|
| Lipophilicity (logD) | logD ≤ 5 [6] | Can be higher, but requires balancing solubility | Increases permeability, but can decrease solubility and increase efflux risk [5] [6] |
| Molecular Weight (MW) | MW ≤ 500 [6] | Neutral compds: up to ~700; Charged compds: ~400-500 [5] | Higher MW generally decreases passive permeability [5] |
| Hydrogen Bond Donors (HBD) | HBD ≤ 5 [6] | Can be higher with IMHB formation [8] | More HBDs significantly reduce permeability [5] |
| Hydrogen Bond Acceptors (HBA) | HBA ≤ 10 [6] | Can be higher with IMHB formation [8] | More HBAs reduce permeability [5] |
| Polar Surface Area (PSA) | PSA ≤ 140 Ų [6] | Can be higher with conformational flexibility/IMHB [6] | Inverse relationship with permeability [6] |
| Intramolecular H-Bonds | Can improve permeability [7] | Critical for shielding polarity and enabling permeability [8] [6] | Shields H-bonding groups, increasing effective lipophilicity [7] |
Issue 2: Inconsistent or Irreproducible Results in Caco-2 Permeability Models
Potential Causes and Solutions:
Cause: Variable Cell Monolayer Integrity.
- Solution: Routinely monitor Transepithelial Electrical Resistance (TEER) to ensure the formation of confluent, differentiated monolayers with intact tight junctions before initiating transport studies [10].
- Solution: Avoid overcrowding and dome formation by subculturing cells before they reach high confluence (e.g., ~50% instead of 80%) [10].
Cause: Instability of Cell Line Characteristics Over Time.
- Solution: Limit continuous cell cultures to a defined number of passages (e.g., three months) to prevent genetic drift and changes in phenotype, gene expression, and transporter function that can alter permeability [10].
Cause: Solvent (DMSO) Interference.
- Solution: Perform solvent tolerance tests. Keep DMSO concentrations consistent and as low as possible in assay buffers, as it can affect cell growth, protein stability, and compound binding [10].
Experimental Protocols
Protocol 1: Determining Intracellular Bioavailability (Fic)
Methodology Cited: Prediction of intracellular exposure bridges the gap between target- and cell-based assays [1].
1. Principle: This label-free method quantifies the fraction of extracellularly added compound that is bioavailable inside the cell in its unbound form (Fic). It combines measurements of total cellular compound accumulation and the intracellular unbound fraction.
2. Workflow: The following diagram illustrates the key steps in determining Fic.
3. Key Steps:
- Cell Incubation: Incubate the relevant cell type (e.g., PBMCs, HeLa cells) with the test compound at a physiologically relevant concentration and temperature [1] [9].
- Separation and Washing: Rapidly separate the cells from the medium by centrifugation through an oil layer or rapid filtration, followed by a gentle wash to remove residual extracellular compound [1].
- Quantification: Lyse the cell pellet and quantify the total intracellular compound concentration using a sensitive mass spectrometry method (e.g., RapidFire-MS for higher throughput or UHPLC-MS/MS) [1] [9].
- Unbound Fraction: Determine the unbound fraction (fu,cell) in the cell lysate using methods like equilibrium dialysis.
- Calculation: Calculate Fic as the product of the cellular-to-medium concentration ratio (Kp) and the unbound fraction in the cell (fu,cell) [1].
Protocol 2: Investigating the Impact of Intramolecular Hydrogen Bonds
Methodology Cited: Impact of Stereospecific Intramolecular Hydrogen Bonding on Cell Permeability [8].
1. Principle: Compare the permeability, lipophilicity (LogD), and pKa of stereoisomers that have the potential to form intramolecular hydrogen bonds. A measurable difference in these properties, particularly a lower measured pKa and higher LogD for the isomer capable of forming the IMHB, provides evidence of its formation and functional impact.
2. Key Steps:
- Stereoisomer Synthesis/Sourcing: Obtain a series of stereoisomers (e.g., cis/trans diastereomers) of the compound of interest.
- Physicochemical Profiling: Experimentally measure the key properties for all stereoisomers:
- Lipophilicity: Determine LogD at pH 7.4.
- Permeability: Use a cell-based model like Caco-2 or MDCK.
- Acidity Constant: Determine pKa, particularly for the involved basic amine or acidic group.
- Computational and Spectroscopic Analysis:
- Use molecular mechanics calculations (e.g., with continuum solvation models) to identify low-energy conformations and estimate the strain energy difference between neutral and protonated forms. The neutral form of the isomer capable of forming a strong IMHB will often be stabilized [8].
- Confirm the presence of IMHB using NMR spectroscopy [8].
3. Interpretation: Isomers that form stable IMHBs will typically display a lower measured pKa (because the neutral form is stabilized), a higher LogD (due to reduced apparent polarity), and consequently, higher passive cell permeability compared to isomers that cannot form the bond [8].
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Permeability and Intracellular Exposure Research
| Item / Reagent | Function / Application | Key Considerations |
|---|---|---|
| Caco-2 Cells | In vitro model for predicting intestinal drug permeability and absorption. Forms confluent monolayers with tight junctions [10]. | Monitor passage number and culture time to prevent phenotype drift. Use TEER to validate monolayer integrity [10]. |
| Madin-Darby Canine Kidney (MDCK) Cells | Alternative cell line for permeability screening. Often has lower endogenous transporter expression than Caco-2 cells [6]. | Useful for assessing passive transcellular permeability with less interference from efflux transporters. |
| Parallel Artificial Membrane Permeability Assay (PAMPA) | Non-cell-based, high-throughput assay to measure passive diffusion potential across an artificial membrane [1] [6]. | Does not account for active transport or metabolism. Can have a poor correlation with intracellular bioavailability (Fic) [1]. |
| Cyclosporine A | Pan-inhibitor of efflux transporters (e.g., P-glycoprotein). Used in transport assays to investigate the role of active efflux [1]. | A significant increase in permeability in its presence indicates the compound is an efflux transporter substrate. |
| Liquid Chromatography-Mass Spectrometry (LC-MS/MS) | Gold-standard technique for sensitive and specific quantification of drug concentrations in biological matrices like cell lysates [9]. | Enables direct measurement of intracellular compound concentration. RapidFire-MS systems can significantly increase throughput [9]. |
| Transepithelial Electrical Resistance (TEER) Meter | Instrument to measure the electrical resistance across a cellular monolayer. A high TEER value indicates intact tight junctions and a valid model for permeability studies [10]. | Critical for quality control of Caco-2 and other epithelial cell models before and during permeability experiments [10]. |
FAQs: Navigating Permeability in Intracellular Target Assays
Why do my bRo5 compounds (like PROTACs or macrocyclic peptides) show poor permeability in standard Caco-2 assays, and how can I improve data quality? Standard permeability assays often fail with bRo5 compounds due to technical limitations like poor recovery, low detection sensitivity, and nonspecific binding [11]. An optimized "equilibrated" Caco-2 assay closes this gap. Key modifications include [11]:
- Pre-incubation Step: A 60-90 minute pre-incubation of the compound with the cell monolayer before the main permeability measurement.
- Extended Incubation Time: Allows measurement closer to equilibrium, which is critical for very low-permeability compounds.
- Additive in Buffer: Using Hank's Balanced Salt Solution (HBSS) with 1% Bovine Serum Albumin (BSA) to reduce nonspecific binding.
- Optimized LC-MS/MS Analytics: Enhances detection sensitivity for compounds with low permeability.
This optimized assay successfully characterized over 90% of tested compounds, most of which were bRo5 (68%) and could not be measured using the standard protocol [11].
What are the key molecular properties to focus on when designing permeable bRo5 compounds? While bRo5 compounds violate traditional rules, analysis of successful oral macrocyclic drugs reveals new, practical guidelines. For orally bioavailable macrocycles, key thresholds are [12]:
- Hydrogen Bond Donors (HBD) ≤ 7
- Meet at least one of the following:
- Molecular Weight (MW) < 1,000 Da
- Calculated logP (cLogP) < 6
- Topological Polar Surface Area (TPSA) < 180 Ų
For de novo designed macrocycles, a more restrictive guideline of amide-type HBDs ≤ 2 is recommended [12]. The concept of "chameleonicity" – a molecule's ability to shield polar groups in lipid membranes and expose them in aqueous environments – is also critical for bRo5 permeability [13].
Which computational models are most reliable for predicting cyclic peptide permeability? A comprehensive benchmark of 13 AI methods found that graph-based models, particularly the Directed Message Passing Neural Network (DMPNN), consistently achieve top performance for predicting cyclic peptide membrane permeability [14]. Furthermore, the Cyclic Peptide Membrane Permeability (CPMP) model, based on a Molecular Attention Transformer, has demonstrated robust performance, outperforming traditional machine learning methods across several metrics [15].
Table 1: Permeability Classification and Predictive Cut-offs from Optimized Caco-2 Assay [11]
| Absorption Classification | Permeability (Papp) Cut-off (10⁻⁶ cm/s) | Efflux Ratio (ER) Cut-off |
|---|---|---|
| High Absorption | > 1.5 | < 2.5 |
| Moderate Absorption | 0.5 - 1.5 | 2.5 - 4.0 |
| Low Absorption | < 0.5 | > 4.0 |
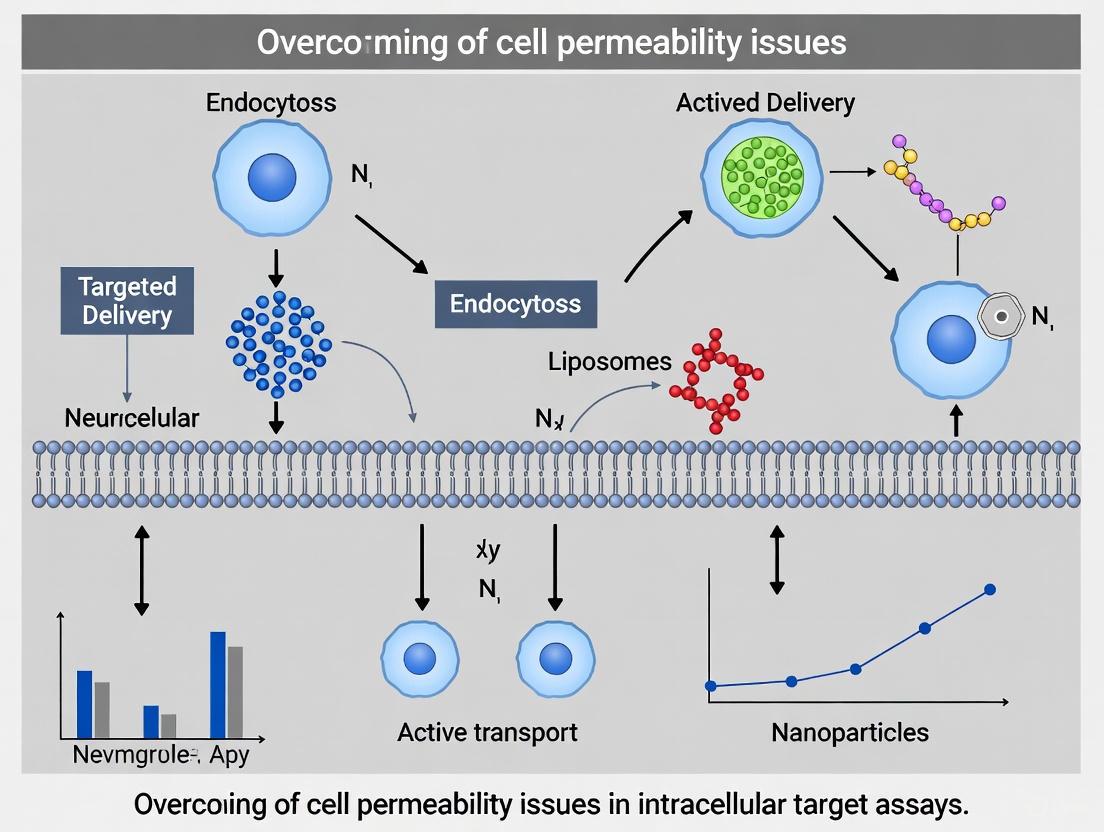
How can the prodrug strategy overcome permeability challenges for bRo5 compounds? The prodrug approach is a versatile strategy for enhancing membrane permeability. Prodrugs are inactive derivatives designed to release the active parent drug after enzymatic or chemical transformation in vivo [16]. This strategy can significantly improve permeability by [16]:
- Masking Polar Groups: Temporarily converting polar ionizable groups (e.g., carboxylic acids, amines) into less polar, more lipophilic promoieties (e.g., esters, amides) to enhance passive diffusion.
- Utilizing Transporters: Designing prodrugs that are substrates for active influx transporters in the intestinal membrane.
- Optimizing PROTACs: Applying prodrug technology to PROteolysis TArgeting Chimeras (PROTACs) through conjugation to improve their cell permeability and overall oral bioavailability.
Experimental Protocols
Key Materials:
- Cell Line: Caco-2 cells (assay-ready frozen format)
- Plates: 0.4 µm Millicell 96-well transwell plates
- Buffers: HBSS (pH 7.4), with and without 1% (w/v) BSA
- Compound Solution: Test compound at 1-3 µM in HBSS with 1% BSA and monolayer-integrity marker (e.g., 80 µM lucifer yellow)
- Analytical Instrument: LC-MS/MS system
Procedure:
- Cell Culture: Seed Caco-2 cells at a density of 40,000 cells per well on transwell inserts. Culture for 7-8 days with medium changes to ensure monolayer differentiation and integrity.
- Pre-incubation (Equilibration Step):
- Add compound solution to the donor compartment.
- Fill the receiver compartment with HBSS buffer containing 1% BSA.
- Incubate for 60-90 minutes at 37°C, 5% CO₂.
- Main Incubation:
- Remove the pre-incubation solution.
- Rinse the cells with HBSS containing 1% BSA.
- Add fresh compound solution to the donor compartment and fresh receiver buffer to the receiver compartment.
- Incubate for 60 minutes at 37°C, 5% CO₂.
- Sample Collection and Analysis:
- Collect samples from both donor and receiver compartments.
- Quench samples with a solution (e.g., 30% acetonitrile with internal standard).
- Analyze compound concentrations using LC-MS/MS.
- Data Calculation:
- Calculate apparent permeability (Papp) using the standard equation.
- Calculate Efflux Ratio (ER) as Papp(B-A) / Papp(A-B).
Key Materials:
- Software/Code: Access to model implementations (e.g., CPMP from GitHub, or other graph-based models like DMPNN).
- Input Data: Molecular structures of compounds in SMILES notation.
- Computing Environment: Python environment with necessary deep learning libraries (e.g., PyTorch, TensorFlow).
Procedure for CPMP Model [15]:
- Data Preprocessing:
- Input cyclic peptide structures as SMILES strings.
- Generate 3D molecular conformations and calculate molecular features (atom features, bond information, inter-atomic distances).
- Model Inference:
- The Molecular Attention Transformer (MAT) architecture processes the input matrices (distance, adjacency, atom features).
- The integrated attention mechanism and feed-forward networks generate a predicted permeability value (LogPexp).
- Output and Interpretation:
- The model outputs a continuous permeability value.
- Compare the predicted value against established benchmarks (e.g., Table 1) to assess permeability potential.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for Permeability Studies of bRo5 Compounds
| Reagent / Material | Function / Application | Key Considerations |
|---|---|---|
| Caco-2 Cell Line | A human colon adenocarcinoma cell line that forms polarized monolayers, simulating the human intestinal epithelium for permeability screening. | Use assay-ready, characterized cells for reproducibility. Extended differentiation time (7-21 days) required [17] [11]. |
| Transwell Plates (0.4 µm) | Permeable supports for growing cell monolayers; allow for bidirectional transport studies. | Critical for assessing apical-to-basolateral and basolateral-to-apical flux to calculate Efflux Ratio [11]. |
| Bovine Serum Albumin (BSA) | Additive to assay buffers to reduce nonspecific binding of lipophilic bRo5 compounds to plastic and cells. | Using 1% BSA in HBSS significantly improves compound recovery in the equilibrated Caco-2 assay [11]. |
| LC-MS/MS System | Highly sensitive analytical instrument for quantifying low concentrations of permeated compounds. | Essential for detecting the low permeation levels typical of bRo5 compounds [11]. |
| PAMPA Kit | Parallel Artificial Membrane Permeability Assay; a high-throughput, cell-free system for estimating passive permeability. | Useful for early-stage screening; lacks biological transporters present in cellular models [17]. |
| PROTAC & Macrocycle pKa Dataset | Curated experimental data used to train and validate predictive software algorithms. | Improves accuracy of in silico pKa predictions for complex molecules, a key parameter affecting permeability [18]. |
Decision Pathways and Workflows
From Traditional Assays to Cutting-Edge Models: A Toolkit for Permeability Assessment
Troubleshooting Guides and FAQs
Frequently Asked Questions
Q1: My high-throughput screening results from PAMPA do not match my later Caco-2 data. Which result should I trust?
This is a common issue stemming from the fundamental differences between these models. PAMPA measures pure passive transcellular permeability through an artificial membrane [19]. If your compound is affected by active transport, efflux, or paracellular pathways, Caco-2 data will provide a more physiologically relevant picture [20]. You should trust the Caco-2 data for final decision-making regarding intestinal absorption, as it incorporates more biological complexity. Use PAMPA for early-stage rank-ordering of compounds based on passive permeability [19].
Q2: Why is my compound's apparent permeability (Papp) so low, even though computer models predict high membrane permeability?
The most likely cause is a dominant aqueous boundary layer (ABL) effect. The apparent permeability (Papp) you measure is a composite value influenced by multiple resistances in series. For compounds with high intrinsic membrane permeability, the rate-limiting step is often diffusion through the unstirred water layers adjacent to the cellular monolayer, not the membrane itself [21] [22]. A meta-analysis of literature data found that about half of all published Papp values are limited by this diffusion through aqueous layers rather than by the membrane [21]. To diagnose this, you can perform experiments at different stirring speeds; if Papp increases with stirring speed, your measurement is ABL-limited.
Q3: When should I choose MDCK-MDR1 cells over standard Caco-2 cells?
MDCK-MDR1 cells are particularly advantageous when you need to rapidly assess the interaction of your compound with the P-glycoprotein (P-gp) efflux transporter [20]. They are genetically modified to overexpress human P-gp, providing a sensitive system for identifying P-gp substrates. Furthermore, MDCK cells form tight monolayers in just 3-5 days, compared to the 21-day differentiation period required for Caco-2 cells, making them suitable for higher-throughput studies [20]. Choose Caco-2 cells when you need a more comprehensive model that expresses a wider array of native human transporters and enzymes.
Q4: What are the key factors to consider when interpreting permeability data for intracellular target assays?
For intracellular targets, the therapeutic must not only cross the intestinal epithelium (measured by Caco-2/PAMPA) but also the plasma membrane of the target cell [23] [24]. Be aware that:
- Efflux Transporters: Activity in gut epithelium (e.g., by P-gp) can limit overall bioavailability, but may not affect cellular uptake in tissues with lower transporter expression.
- Physicochemical Properties: Deviations from traditional rules (e.g., Lipinski's Rule of 5) are common for non-oral modalities or newer therapeutic classes like macrocyclic peptides and metallocomplexes [23] [24]. Strategies like vectorization with cell-penetrating peptides (CPPs) can be employed to overcome poor permeability [24].
Troubleshooting Common Experimental Issues
Problem: High Variability in Replicate Permeability Measurements
- Potential Cause 1: Inconsistent cell culture conditions or monolayer integrity.
- Solution: Strictly standardize culture protocols. For Caco-2 cells, ensure a consistent and full 21-day differentiation period. Before each experiment, validate monolayer integrity by measuring Transepithelial Electrical Resistance (TEER) or using a standard paracellular marker like Lucifer Yellow [20].
- Potential Cause 2: Compound precipitation or non-specific binding to equipment.
- Solution: Check the solubility of your compound in the assay buffer. Consider using a low concentration of a co-solvent like DMSO (e.g., 0.5-1%), and ensure proper calibration and cleaning of instrumentation [19].
- Potential Cause 3: Instability or metabolism of the compound during the assay.
- Solution: Analyze samples post-assay (e.g., by LC-MS) to confirm the integrity of the parent compound. Low recovery can indicate compound adsorption, degradation, or metabolism [21].
Problem: Low Compound Recovery in the Permeability Assay
- Potential Cause 1: Significant non-specific binding to the plastic plate or filter.
- Solution: Use surface-modified plates or include a blocking agent. Calculate mass balance; recovery should ideally be between 90-110%. Include control compounds with known recovery profiles [19].
- Potential Cause 2: Compound is a substrate for efflux transporters or is metabolized by the cells.
Problem: Poor Correlation Between In Vitro Permeability and In Vivo Oral Absorption
- Potential Cause 1: The in vitro assay conditions do not adequately mimic the in vivo pH environment.
- Solution: The pH-partition hypothesis states that permeability can vary with pH. Utilize PAMPA or other assays at multiple pH values (e.g., pH 5.0 for the duodenum and pH 7.4 for the distal intestine and systemic circulation) to build a more predictive absorption profile [19].
- Potential Cause 2: overlooking the role of paracellular transport.
- Solution: The mechanistic model of Bittermann and Goss highlights that for many small, polar compounds, the paracellular pathway can be dominant [22]. Analyze your compound's molecular weight and polarity to evaluate if this pathway is significant.
Data Presentation and Comparison
Comparison of Key Permeability Models
Table 1: Characteristics and Applications of Caco-2, MDCK, and PAMPA Assays
| Feature | Caco-2 Model | MDCK-MDR1 Model | PAMPA |
|---|---|---|---|
| Origin/Type | Human colorectal adenocarcinoma cell line [20] | Canine kidney cell line, genetically modified [20] | Artificial membrane (non-cellular) [19] |
| Culture Time | ~21 days for full differentiation [20] | 3-5 days [20] | Not applicable |
| Key Transport Mechanisms | Passive transcellular, paracellular, active influx/efflux, metabolism [20] [22] | Passive transcellular, dominant P-gp efflux [20] | Passive transcellular diffusion only [19] |
| Primary Application | Gold standard for predicting human intestinal absorption [22] | Rapid screening for P-gp efflux and permeability [20] | High-throughput rank-ordering of passive permeability in early discovery [19] |
| Throughput | Low to medium | Medium | High |
| Data Output | Apparent Permeability (Papp), efflux ratio | Apparent Permeability (Papp), efflux ratio | Effective Permeability (Peff) [19] |
| Major Limitations | Long culture time, batch-to-batch variability, complex transport mechanisms [19] [20] | Less physiologically relevant than Caco-2; does not model full array of human transporters [20] | Cannot model active transport, paracellular transport, or metabolism [19] |
Table 2: Quantitative Permeability Ranges and Data Reliability Considerations
| Parameter | Typical Range/Value | Context and Limitation |
|---|---|---|
| PAMPA Peff (pH 5) | Low: < 10 x 10⁻⁶ cm/s; Moderate/High: > 10 x 10⁻⁶ cm/s [19] | NCATS uses this cutoff. Correlates ~85% with in vivo oral bioavailability in rats/mice [19]. |
| Papp Measurement Range | Log Papp ~ -8 to -4 log cm/s [25] | Cell-based methods are physically limited to this range, restricting data for highly permeable compounds [25]. |
| Data Reliability | Only ~25% of published Papp values yield reliable intrinsic permeability (P₀) [21] | A study of 318 compounds found ~50% were limited by aqueous boundary layers, others by paracellular transport or recovery issues [21]. |
Experimental Protocols
Detailed Protocol: Double-Sink PAMPA at pH 5
This protocol is adapted from the high-throughput method used at NCATS for early passive permeability screening [19].
1. Principle: The Parallel Artificial Membrane Permeability Assay (PAMPA) measures passive diffusion of a compound through a proprietary gut-inspired (GIT-0) lipid immobilized on a filter, which separates a donor compartment (pH 5.0) from an acceptor compartment (pH 7.4). The "double-sink" condition in the acceptor well helps maintain a concentration gradient, mimicking in vivo conditions.
2. Reagents and Materials:
- Test Compound: 10 mM stock in DMSO.
- Buffers: PRISMA HT buffer (pH 5.0) for donor wells; Acceptor Sink Buffer (pH 7.4).
- Lipid: GIT-0 lipid (proprietary to Pion Inc.).
- Equipment: 96-well stirwell sandwich plates with stirrers; Gutbox for stirring; UV plate reader or UPLC-MS for quantification.
- Controls: High-permeability control (e.g., Verapamil); low-permeability control (e.g., Ranitidine).
3. Procedure: 1. Plate Preparation: Immobilize the GIT-0 lipid on the filter of the acceptor plate. 2. Sample Preparation: Dilute the test compound from the 10 mM DMSO stock to 0.05 mM in pH 5.0 PRISMA HT buffer. The final DMSO concentration should be ≤ 0.5% (v/v). 3. Loading: Add the compound solution to the donor wells. Add the acceptor sink buffer (pH 7.4) to the acceptor wells. 4. Incubation: Assemble the sandwich plate and incubate at room temperature for 30 minutes with stirring in a Gutbox (to reduce the aqueous boundary layer). 5. Analysis: Measure the concentration of the test article in both donor and acceptor compartments using a UV plate reader. If UV detection is unsuitable, use a validated UPLC-MS method. 6. Calculation: The effective permeability (Peff in 10⁻⁶ cm/s) is calculated by the Pion software using the flux data from both compartments.
Detailed Protocol: Caco-2 Permeability Assay
This protocol outlines the standard procedure for assessing permeability and transport mechanisms in differentiated Caco-2 cell monolayers [20] [22].
1. Principle: Caco-2 cells are grown on a porous filter until they differentiate into an enterocyte-like monolayer with tight junctions. The permeability of a compound from the apical (A) to basolateral (B) side and vice versa is measured to determine apparent permeability (Papp) and identify potential efflux transporter involvement.
2. Reagents and Materials:
- Cell Line: Caco-2 cells (use a consistent, low-passage source).
- Culture Media: Dulbecco's Modified Eagle Medium (DMEM) with supplements (e.g., FBS, non-essential amino acids, glutamine).
- Assay Buffers: HBSS or other transport buffers, typically at pH 6.5 apical / 7.4 basolateral to simulate intestinal gradients.
- Transwell Plates: 12 or 24-well plates with polycarbonate filters (e.g., 0.4 µm or 3.0 µm pore size).
- Instrumentation: TEER meter; LC-MS/MS or HPLC for quantitative analysis.
3. Procedure: 1. Cell Culture and Seeding: Seed Caco-2 cells at a high density (~100,000 cells/cm²) onto the apical side of the Transwell filter. Change the media every 2-3 days. 2. Monolayer Differentiation and Integrity Check: Culture the cells for 21 days. Monitor Transepithelial Electrical Resistance (TEER) regularly. Before the experiment, confirm TEER values are acceptably high (e.g., >300 Ω·cm²) for your lab standard. 3. Experiment Setup: * Wash the monolayers with pre-warmed transport buffer. * Add the test compound (typically 5-100 µM) to the donor compartment (for A-B transport, add to apical side; for B-A, add to basolateral side). * Add fresh buffer to the receiver compartment. * Incubate in a shaking incubator (e.g., 37°C, ~150 rpm) for a set time (e.g., 60-120 minutes). 4. Sample Collection: At designated time points, take samples from both donor and receiver compartments. 5. Analysis: Quantify compound concentrations in all samples using LC-MS/MS. Calculate Papp using the formula: Papp = (dQ/dt) / (A * C₀), where dQ/dt is the flux rate, A is the filter surface area, and C₀ is the initial donor concentration. 6. Efflux Ratio Calculation: Efflux Ratio = Papp (B-A) / Papp (A-B). A ratio >2 suggests active efflux.
Signaling Pathways and Experimental Workflows
Diagram: Mechanistic Pathways of Permeation in Cell Monolayers
The following diagram illustrates the three parallel permeation pathways a solute may take through a Caco-2 or MDCK monolayer, as described in mechanistic models [22].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Permeability Assays
| Item | Function/Description | Example Use Case |
|---|---|---|
| GIT-0 Lipid | A proprietary lipid blend optimized to predict gastrointestinal tract passive permeability [19]. | Used in the double-sink PAMPA assay to create a biomimetic artificial membrane. |
| Caco-2 Cell Line | A human colorectal adenocarcinoma cell line that spontaneously differentiates into enterocyte-like cells, forming a polarized monolayer with tight junctions [20]. | The gold-standard cellular model for predicting human intestinal absorption and studying transporter effects. |
| MDCK-MDR1 Cell Line | Madin-Darby Canine Kidney cells genetically modified to stably overexpress the human P-glycoprotein (MDR1) efflux transporter [20]. | A rapid, sensitive model specifically for assessing compound efflux by P-gp. |
| Transwell Plates | Multi-well plates featuring a suspended, porous filter membrane on which cell monolayers are grown [22]. | The physical scaffold for culturing Caco-2 and MDCK cells to separate apical and basolateral compartments in permeability assays. |
| Transepithelial Electrical Resistance (TEER) Meter | An instrument that applies a small alternating current to measure the electrical resistance across a cell monolayer, a key indicator of its integrity and tight junction formation [20]. | Used to validate the quality and confluency of Caco-2 and MDCK monolayers before and after permeability experiments. |
Technical Support Center: FAQs & Troubleshooting Guides
Frequently Asked Questions (FAQs)
Q1: What are the main advantages of using organ-on-a-chip (OoC) platforms over conventional 2D cell cultures for permeability and drug absorption studies?
- A1: Organ-on-a-chip systems offer several critical advantages [26] [27]:
- Physiological Relevance: OoCs mimic organ-level functions and 3D tissue architecture better than 2D monolayers.
- Dynamic Microenvironment: They emulate crucial physiological parameters like fluid perfusion (mimicking blood flow), mechanical forces (e.g., cyclic stretching for lungs or intestines), and tissue-tissue interfaces [26] [28].
- Improved Predictivity: The enhanced physiological context leads to more accurate predictions of drug absorption, toxicity, and efficacy, potentially reducing the reliance on misleading animal models [26].
- Automation and Reduction: They reduce labor costs, human error, and reagent use through automated operation in miniaturized systems [26].
Q2: How can I improve the maturation and functionality of organoids in my culture system?
- A2: Achieving mature, functional organoids often requires moving beyond static cultures. Key strategies include [26] [28]:
- Integration with Microfluidics: Culturing organoids within a microfluidic chip (creating an organoid-on-a-chip) provides precise control over the biochemical and biophysical microenvironment, leading to better differentiation and function [26] [28].
- Environmental Control: Providing dynamic fluid flow improves nutrient supply and waste removal, preventing necrosis in the organoid core.
- Mechanical Stimulation: Incorporating physiological cues like fluid shear stress or stretching can drive organ-specific maturation, such as in gut or lung models [28].
Q3: My model shows low correlation with in vivo human data for drug permeability. What could be wrong?
- A3: Low correlation often stems from an oversimplified in vitro model. Consider these enhancements [17]:
- Model Complexity: Transition from simple monocultures (e.g., Caco-2 alone) to more complex co-cultures. For instance, co-culturing Caco-2 with mucus-producing HT29-MTX cells better replicates the intestinal barrier by including a mucin layer.
- Dynamic Flow: Implement perfusion using OoC systems to mimic the in vivo environment where flowing fluid affects compound absorption and cell behavior.
- Source Cells: Utilize patient-derived or induced pluripotent stem cell (iPSC)-derived organoids to incorporate human genetic background and specific disease phenotypes [26] [28].
Q4: What are the key regulatory changes supporting the use of these advanced in vitro models?
- A4: There is significant regulatory momentum to accept human biology-based test methods [26]:
- US FDA Modernization Act (2021): This act allows drug manufacturers to use alternative methods, including "cell-based assays, organ chips and microphysiological systems," instead of animal testing for safety and efficacy data.
- European Parliament Resolution (2021): A resolution was adopted to actively reduce and replace procedures with live animals in research, regulatory testing, and education.
Troubleshooting Guides
Problem: High Background Signal or Non-Specific Staining in Intracellular Imaging or Flow Cytometry
| Possible Cause | Recommendation |
|---|---|
| Insufficient Blocking | Block cells with Bovine Serum Albumin, Fc receptor blocking reagents, or normal serum from the same host as your antibodies prior to staining [29]. |
| Presence of Dead Cells | Use a viability dye (e.g., PI, 7-AAD, or a fixable viability dye) to gate out dead cells during analysis [29]. |
| Antibody Concentration Too High | Titrate your antibodies to find the optimal concentration. Avoid using too much antibody [29]. |
| Incomplete Washing | Perform additional wash steps between antibody incubations and after the final staining step to remove unbound reagents [29]. |
Problem: Weak or No Fluorescence Signal in Flow Cytometry
| Possible Cause | Recommendation |
|---|---|
| Inadequate Fixation/Permeabilization | For intracellular targets, ensure you use a validated protocol. Use fresh, ice-cold methanol added drop-wise while vortexing for homogeneous permeabilization [29]. |
| Dim Fluorochrome for Low-Density Target | Always pair your lowest-density target (e.g., a sparsely expressed protein) with the brightest fluorochrome (e.g., PE). Use dimmer fluorochromes (e.g., FITC) for high-density targets [29]. |
| Incorrect Instrument Settings | Ensure the cytometer's laser and PMT settings are configured for the excitation and emission wavelengths of the fluorochromes you are using [29]. |
| Clogged Flow Cell | Follow the manufacturer's instructions to unclog the system, often by running 10% bleach followed by distilled water [29]. |
Problem: Poor Cell Permeability for a Novel Compound in our Intestinal Barrier Model
| Possible Cause | Recommendation |
|---|---|
| Overly Simple Model | Use a co-culture of Caco-2 and HT29-MTX cells to incorporate a physiologically relevant mucus barrier, which can significantly impact compound permeation [17]. |
| Lack of Physiological Transporters | Characterize the expression of key influx/efflux transporters (e.g., P-gp) in your model. Consider using iPSC-derived enterocytes for a more complete transporter profile [17]. |
| Static Culture Conditions | Transition to an organ-on-a-chip model with continuous perfusion to maintain a healthy, differentiated barrier and mimic the in vivo flow conditions that influence permeability [28] [17]. |
Detailed Protocol: Establishing a Gut-on-a-Chip for Permeability Studies
This protocol outlines the creation of a human gut-on-a-chip model that recapitulates the intestinal barrier for assessing drug and compound permeability [28] [27].
Key Materials (Research Reagent Solutions)
| Item | Function |
|---|---|
| Microfluidic Chip | The physical device, often made of PDMS, containing microchannels and chambers to house cells and perfuse media [27]. |
| Extracellular Matrix (ECM) | A scaffold, such as Matrigel or collagen, upon which cells are seeded to support 3D growth and organization [28]. |
| Caco-2 Cells | A human colorectal adenocarcinoma cell line that differentiates into enterocyte-like cells, forming the primary intestinal barrier. |
| HT29-MTX Cells | A mucin-producing cell line. Used in co-culture with Caco-2 to introduce a physiologically relevant mucus layer [17]. |
| Differentiation Media | Cell culture media formulated to promote the differentiation of cells into a mature, functional intestinal epithelium. |
| Peristaltic Pump or Syringe Pump | A device to generate controlled, low-rate fluid flow through the microfluidic channels, mimicking blood flow and intestinal peristalsis [28]. |
Methodology
- Chip Preparation: Sterilize the microfluidic chip (e.g., via UV light or ethanol). Coat the central cell culture chamber with an appropriate ECM (e.g., diluted Matrigel) and incubate to allow gel formation.
- Cell Seeding: Trypsinize and prepare a cell suspension of Caco-2 cells or a co-culture of Caco-2 and HT29-MTX cells (e.g., 90:10 ratio). Introduce the cell suspension into the ECM-coated chamber at a high density to ensure confluency.
- Initial Attachment: Allow the chip to rest in an incubator (37°C, 5% CO2) for a few hours without flow to let the cells attach to the ECM.
- Initiation of Perfusion: After cell attachment, connect the chip to the pump system and begin a slow, continuous perfusion of differentiation media through the channels adjacent to the culture chamber. The flow rate should be low initially (e.g., 10-50 µL/h) and can be gradually increased.
- Maturation and Differentiation: Culture the cells under continuous flow for 10-21 days to allow for full differentiation and formation of a polarized epithelium with tight junctions. The model can be enhanced by applying cyclic mechanical strain (stretching) to mimic intestinal peristalsis if the chip design allows it.
- Permeability Assay: Your gut-on-a-chip model is now ready for experiments. To measure permeability, introduce the test compound to the apical (luminal) inlet channel and collect effluent from the basolateral (blood) outlet channel at timed intervals. Analyze the concentration of the compound that crossed the barrier using methods like HPLC or MS. The apparent permeability (Papp) can be calculated using standard formulas.
The table below summarizes key characteristics of different in vitro models used in permeability and drug development research [26] [17].
| Model System | Physiological Relevance | Throughput | Reproducibility | Key Advantages | Key Limitations |
|---|---|---|---|---|---|
| 2D Monolayer (Caco-2) | Low-Moderate | High | High | Low cost, standardized, high-throughput suitable for early screening. | Lacks 3D architecture, no mucus layer, no dynamic flow, transporter expression can differ from in vivo. |
| 3D Organoids | High | Low | Low-Moderate | Patient-specific, captures complex 3D architecture and cell types, excellent for disease modeling. | Low-throughput, variability between batches, static culture limits nutrient/waste exchange (core necrosis). |
| Organ-on-a-Chip | High | Moderate | Moderate-High | Recapitulates dynamic microenvironment (flow, stretch), high human physiological relevance, allows real-time monitoring. | More complex setup and operation, requires specialized equipment, can be lower throughput than 2D. |
| Multi-Organ-Chip | Very High | Low | Moderate | Studies complex organ-organ interactions, predicts systemic toxicity and metabolite transport. | Highly complex, challenging to balance organ ratios and media requirements, data interpretation can be difficult. |
Visualizing Experimental Workflows
The following diagrams illustrate core concepts and workflows in advanced in vitro systems.
Frequently Asked Questions (FAQs)
FAQ 1: What is the key advantage of using multitask learning over single-task models for permeability prediction? Multitask Learning (MTL) achieves higher predictive accuracy by leveraging shared information across related endpoints, such as different permeability assays and efflux ratios. For instance, a MTL model trained on a harmonized internal dataset of over 10,000 compounds demonstrated superior performance by simultaneously learning from Caco-2, MDCK, and MDCK-MDR1 assay data. This approach allows the model to identify underlying patterns that are common across different but related experimental measures, which single-task models might miss [30].
FAQ 2: Why is my QSPR model for permeability performing poorly on new chemical series? Poor generalization to new chemical scaffolds is often due to the model's limited applicability domain and high experimental variability in the training data. A benchmark study on cyclic peptide permeability highlighted that models evaluated with a scaffold split (which separates compounds based on their core structure during testing) showed substantially lower generalizability compared to a random split. This indicates that many models fail to learn fundamental permeability principles that transfer across diverse chemical structures. Ensuring your training data covers a broad chemical space and using models like Directed Message Passing Neural Networks (DMPNN) that are robust to scaffold changes can mitigate this issue [14].
FAQ 3: Which molecular features are most critical to include for accurate passive permeability prediction? While traditional descriptors like calculated logP (lipophilicity) and Total Polar Surface Area (TPSA) are important, recent studies show that augmenting models with predicted physicochemical properties like pKa and LogD significantly improves accuracy for both passive permeability and efflux endpoints. For example, a study using message-passing neural networks (MPNNs) found that including these features provided a notable performance boost [30]. However, for more complex molecules like cyclic peptides, graph-based models that learn directly from the molecular structure often outperform those relying solely on pre-defined descriptors [14].
FAQ 4: How does intracellular bioavailability (Fic) differ from membrane permeability, and why is it important? Intracellular bioavailability (Fic) measures the fraction of an unbound drug that is available inside the cell to engage with its target, providing a direct link to cellular potency. In contrast, standard membrane permeability assays (like PAMPA) measure the rate of transport across a membrane. Research has shown that Fic is a net result of permeability, active transport, metabolism, and nonspecific binding. A study on p38α inhibitors found a poor correlation between Fic and PAMPA permeability, and Fic was more effective at explaining the observed drop in cellular potency compared to biochemical assays. Measuring Fic in pharmacologically relevant cell types provides a more accurate prediction of a compound's intracellular efficacy [1].
FAQ 5: When should I use molecular dynamics simulations versus machine learning for permeability prediction? The choice depends on your goal, the number of compounds, and available computational resources. Table 1 summarizes the characteristics of different computational approaches.
Table 1: Comparison of Computational Approaches for Permeability Prediction
| Method | Key Principle | Typical Use Case | Relative Computational Cost |
|---|---|---|---|
| Lipophilicity Relations (QSPR) | Relates logP and other simple descriptors to permeability [31]. | High-throughput virtual screening of large compound libraries. | Low |
| Machine Learning (e.g., GNNs, Random Forest) | Learns complex structure-permeability relationships from large datasets [30] [14]. | Accurate prediction for drug-like molecules within a model's applicability domain. | Medium |
| Molecular Dynamics (MD) Simulations | Uses physics-based models to simulate molecule movement through a lipid bilayer [31] [32]. | Mechanistic studies and predictions for novel or complex molecules (e.g., cyclic peptides). | High |
FAQ 6: My compound is potent in a biochemical assay but inactive in a cellular assay. Could permeability be the issue? Yes, this "cell drop-off" is a common issue in drug discovery and is frequently linked to poor intracellular target exposure. A study on MAPK14 (p38α) inhibitors found that while biochemical and cellular pIC50 correlated well, compounds were on average ten times less potent in the cellular assay. This drop-off was explained by low intracellular bioavailability (Fic). By correcting the biochemical potency with the measured Fic, researchers could accurately predict the cellular potency, confirming that insufficient permeation was the primary cause [1].
Troubleshooting Guides
Issue 1: Handling High Experimental Variability in Permeability Data
Problem: Your computational model is inconsistent and you suspect noisy training data from different laboratory sources is the cause.
Background: The accuracy of QSPR models is heavily dependent on the consistency of the underlying experimental data. Apparent permeability coefficient (Papp) values for the same compound can vary significantly due to differences in experimental protocols (e.g., pH, use of inhibitors, cell passage number) [3].
Solution:
- Data Curation: Prioritize data generated from a single, consistent laboratory protocol. If using public data, meticulously curate it by:
- Grouping data by assay type (e.g., separate Caco-2 from PAMPA data).
- Noting critical experimental conditions (e.g., pH gradient, transporter inhibitors) and filtering for consistency [3].
- Internal Standardization: Always include a set of reference compounds with well-established permeability values in your experimental assays. This allows for normalization of your data and makes it more comparable across different batches and studies [3].
- Modeling Strategy: If diverse data sources must be used, consider a multitask learning approach where the assay condition or laboratory source is treated as a separate but related task. This can help the model learn to distinguish between true permeability and experimental noise [30].
Issue 2: Improving Model Generalizability to Novel Chemotypes
Problem: Your model performs well on test compounds similar to its training set but fails on new chemical scaffolds.
Background: This is a classic problem of a model operating outside its "applicability domain." It often occurs when the training data lacks sufficient chemical diversity or the model architecture cannot capture relevant features for new scaffold types [14].
Solution:
- Evaluate with Scaffold Split: During model development and validation, always use a scaffold-based data splitting strategy in addition to random splitting. This provides a more realistic and rigorous assessment of the model's ability to generalize to truly new chemotypes [14].
- Select a Robust Model Architecture: Choose modern graph-based neural networks like Directed Message Passing Neural Networks (DMPNN) or other Graph Neural Networks (GNNs). Benchmark studies have consistently shown their superior performance in permeability prediction for both small molecules and cyclic peptides, especially when generalizing to new scaffolds [30] [14].
- Data Augmentation: For cyclic peptides, data augmentation techniques that generate synthetic but plausible permeability data can help improve feature extraction and model robustness, though their benefit may be limited for highly diverse scaffolds [14].
Issue 3: Predicting Permeability for Large, Flexible Molecules like Cyclic Peptides
Problem: Standard small-molecule permeability models are inaccurate for cyclic peptides.
Background: The permeability of cyclic peptides is governed not only by lipophilicity but also by conformational flexibility and the ability to form internal hydrogen bonds (the "chameleon" property), which are poorly captured by traditional 2D descriptors [14].
Solution:
- Use Specialized Datasets and Models: Train your models on large, curated datasets specific to cyclic peptides, such as the CycPeptMPDB. Formulate the problem as a regression task rather than classification, as regression has been shown to outperform classification for permeability prediction of these molecules [14].
- Leverage Graph Neural Networks: Represent peptides as molecular graphs, as GNNs can learn complex patterns related to molecular size, flexibility, and potential for intramolecular hydrogen bonding that are critical for peptide permeability [14].
- Consider Molecular Dynamics (MD): For critical compounds, use advanced MD workflows like the Orion Permeability Floe. This method uses a weighted-ensemble approach to simulate the path of a molecule through a lipid bilayer, providing not just a permeability coefficient but also kinetic and mechanistic insights into the permeation process, which can guide redesign [32].
Issue 4: Integrating Efflux Transport Prediction into Permeability Assessment
Problem: Your compound shows good passive permeability but is underperforming in cellular assays, potentially due to efflux transporters.
Background: Efflux transporters like P-gp can actively pump compounds out of cells, reducing their effective intracellular concentration. This process is distinct from passive diffusion [30] [1].
Solution:
- Utilize Multitask Models: Implement a multitask model that simultaneously predicts passive permeability (e.g., Caco-2 Papp (a-b)) and efflux ratio (ER) from assays like Caco-2 or MDCK-MDR1. This allows the model to share information between these related endpoints and provide a more comprehensive absorption profile [30].
- Incorporate Mechanistic Assays: Use cell lines transfected with specific human transporters (e.g., MDCK-MDR1) to generate data that explicitly informs on P-gp efflux. Including this data in your models helps pinpoint transporter-specific issues [30].
- Measure Intracellular Bioavailability (Fic): If a compound is a suspected efflux substrate, determine its Fic in the relevant cell type. This metric directly quantifies the intracellular unbound fraction. Furthermore, Fic can be measured in the presence of a pan-inhibitor like cyclosporine A; an increase in Fic confirms active efflux is limiting intracellular exposure [1].
The Scientist's Toolkit
Table 2: Key Research Reagent Solutions for Permeability Research
| Reagent / Resource | Function in Permeability Research |
|---|---|
| Caco-2 Cell Line | A human colorectal adenocarcinoma cell line used to model intestinal drug absorption and study both passive permeability and efflux in the presence of multiple human transporters (e.g., P-gp, BCRP) [30]. |
| MDCK-MDR1 Cell Line | A Madin-Darby canine kidney cell line transfected with the human MDR1 gene. It is specifically used to determine whether a compound is a substrate of the P-glycoprotein (P-gp) efflux transporter [30]. |
| Fluorescein Diacetate (FDA) | A lipophilic fluorescent dye used in cell-based assays to measure membrane integrity and permeability. Non-fluorescent FDA enters cells and is hydrolyzed by esterases to fluorescent fluorescein, which is trapped inside; its uptake or release is measured to quantify permeability changes [33]. |
| Reference Compounds | A set of compounds with well-characterized permeability and efflux properties (e.g., high/low permeability, P-gp substrates/inhibitors). They are essential for standardizing assays and validating computational models across different labs and conditions [3]. |
| Orion Permeability Floe | A commercial software workflow that uses weighted-ensemble molecular dynamics simulations on cloud computing resources to predict passive permeability coefficients and provide kinetic and mechanistic insights for small molecules [32]. |
| CycPeptMPDB Database | A curated public database of cyclic peptide membrane permeability data, compiled from numerous studies. It serves as an essential benchmark dataset for training and validating machine learning models on peptides [14]. |
Experimental Protocols & Workflows
Protocol 1: Benchmarking Machine Learning Models for Permeability Prediction
This protocol is adapted from recent large-scale benchmarking studies [14].
- Data Collection and Curation:
- Obtain a curated dataset such as CycPeptMPDB for cyclic peptides or a harmonized corporate dataset for small molecules [30] [14].
- Standardize molecular structures (e.g., using RDKit) and handle duplicates by either averaging multiple measurements or ensuring all replicates are in the training set to prevent data leakage [30] [14].
- Data Splitting:
- Perform two distinct splitting strategies:
- Random Split: Split the dataset randomly into training, validation, and test sets (e.g., 80:10:10). Repeat this process with multiple random seeds to ensure stability.
- Scaffold Split: Generate Murcko scaffolds for all molecules. Split the data such that the training and test sets contain distinct scaffolds to rigorously assess generalizability [14].
- Perform two distinct splitting strategies:
- Model Training and Evaluation:
- Train a diverse set of models spanning different molecular representations:
- Fingerprint-based: Random Forest, Support Vector Machine.
- Graph-based: Directed Message Passing Neural Network (DMPNN), other Graph Neural Networks.
- Sequence-based: Models using SMILES strings (e.g., RNNs, Transformers).
- For each model and split, evaluate performance using metrics like Root Mean Square Error (RMSE) for regression and Area Under the ROC Curve (ROC-AUC) for classification [14].
- Train a diverse set of models spanning different molecular representations:
The following diagram illustrates the benchmarking workflow.
Protocol 2: Workflow for Integrating Permeability and Efflux Predictions using Multitask Learning
This protocol is based on the methodology described in [30].
- Data Harmonization:
- Compile a dataset containing molecular structures and multiple related endpoints. A key example includes:
- Intrinsic Caco-2 Papp (a-b) (passive permeability)
- Caco-2 Efflux Ratio (ER)
- MDCK-MDR1 ER
- Convert all measurements to a logarithmic scale. Standardize the SMILES representations for all compounds [30].
- Compile a dataset containing molecular structures and multiple related endpoints. A key example includes:
- Feature Engineering and Model Setup:
- Calculate additional molecular features such as pKa and LogD. These can be used to augment the input to the neural network.
- Set up a Multitask Learning (MTL) architecture using a message-passing neural network (MPNN) framework like Chemprop. In this setup, the model has a shared encoder (to learn a general molecular representation) and multiple task-specific output heads (one for each permeability/efflux endpoint) [30].
- Model Training and Validation:
- Train the MTL model and compare its performance against single-task models (STL) on a held-out test set.
- Validate the model's generalizability on an external public dataset to assess its performance beyond the internal data's chemical space [30].
The following diagram illustrates the MTL workflow for permeability prediction.
Celastrol (CeT), a natural pentacyclic triterpenoid isolated from Tripterygium wilfordii, demonstrates significant broad-spectrum therapeutic potential against various diseases, including cancer, inflammatory conditions, and metabolic disorders [34] [35]. Despite its promising bioactivity, the clinical translation of celastrol has been severely hindered by several inherent limitations, primarily its poor aqueous solubility (13.25 ± 0.83 μg/mL at 37°C) and low permeability, which collectively lead to low oral bioavailability (approximately 17.06% in rat models) and significant off-target toxicity [36] [35]. These physicochemical and biopharmaceutical challenges complicate the accurate identification of its intracellular targets, as conventional assays often require sufficient cellular penetration for target engagement. This case study explores the integrated strategies researchers employ to overcome these permeability barriers, enabling successful decoding of celastrol's therapeutic mechanisms and advancing its drug development potential.
Troubleshooting Guides and FAQs
Frequently Asked Questions
Q1: What are the primary factors limiting celastrol's cellular permeability and bioavailability? A1: The primary factors include:
- Poor Aqueous Solubility: Its rigid pentacyclic triterpenoid structure, with a hydrophobic surface and limited polar functional groups, results in very low water solubility [36] [35].
- First-Pass Metabolism: A significant portion of orally administered celastrol is metabolized in the gastrointestinal tract and liver before reaching systemic circulation [36].
- Physicochemical Properties: The molecule's structural features, including intramolecular hydrogen bonding, impede its passive diffusion across cellular membranes [36].
Q2: How can we confirm that celastrol's low efficacy in cellular assays is due to permeability issues and not a lack of target engagement? A2: To diagnose the root cause:
- Compare Activity in Cell-Free vs. Cell-Based Assays: If celastrol shows strong activity in a cell-free enzymatic assay but weak activity in a live cell assay targeting the same pathway, permeability is likely a major limiting factor [37].
- Utilize Permeability Enhancement Tools: Employ strategies like nanocarrier encapsulation or structural modification. A significant increase in cellular efficacy after using these strategies strongly suggests that improving delivery overcame a permeability barrier [36] [35].
- Measure Cellular Uptake Directly: Use analytical techniques like LC-MS/MS to quantify the intracellular concentration of celastrol, providing direct evidence of its poor accumulation [36].
Q3: What are the common pitfalls in flow cytometry when analyzing low-permeability compounds like celastrol, and how can they be addressed? A3: Common issues and solutions include:
- Weak or No Fluorescence Signal:
- Cause: Inadequate cellular uptake of the compound or probe.
- Solution: Optimize fixation and permeabilization protocols. Use ice-cold methanol added drop-wise while vortexing for homogeneous permeabilization. Ensure formaldehyde is methanol-free to prevent premature permeabilization [38].
- High Background Signal:
- Cause: Non-specific binding, often due to the compound's hydrophobicity.
- Solution: Include rigorous controls (unstained, isotype). Block cells with Bovine Serum Albumin or Fc receptor blocking reagents. Perform additional wash steps and titrate antibodies to find the optimal concentration [38].
Q4: Which advanced in vitro models can provide more physiologically relevant permeability data for compounds like celastrol? A4: Beyond traditional monolayer cultures, more advanced models include:
- Microphysiological Systems (MPS/Organs-on-a-Chip): These systems, such as a small airway MPS, recapitulate the 3D tissue architecture, air-liquid interface (ALI), and dynamic fluid flow of human organs, providing a more accurate environment for permeability studies [39] [17].
- Co-culture Models: Co-culturing epithelial cells (e.g., Caco-2) with mucin-producing cells (e.g., HT29-MTX) better mimics the intestinal barrier and can give more predictive permeability data [17].
- 3D Cell Models: Spheroids and organoids derived from induced pluripotent stem cells (iPSCs) offer greater physiological relevance and improved predictability for drug permeability and absorption [17].
Experimental Protocols for Target Identification Despite Permeability Challenges
The following protocols outline a multi-faceted approach to identify targets for impermeable compounds like celastrol, combining direct delivery methods with advanced proteomic techniques.
Protocol 1: Nanoformulation of Celastrol to Enhance Cellular Delivery
Principle: Encapsulating celastrol in nanocarriers improves its aqueous solubility and facilitates its entry into cells, thereby enabling the study of its intracellular interactions [36] [35].
Materials:
- Celastrol (CeT)
- Biocompatible polymer (e.g., PLGA) or lipid (for liposomes)
- Organic solvent (e.g., dichloromethane, acetone)
- Surfactant (e.g., poloxamer, polysorbate 80)
- Probe sonicator
- Magnetic stirrer
- Dialysis membrane or ultrafiltration device
Methodology:
- Nanoparticle Preparation: Dissolve 10 mg of celastrol and 100 mg of PLGA polymer in a suitable organic solvent.
- Emulsification: Add this organic solution drop-wise to an aqueous solution containing 1% (w/v) surfactant while probe-sonicating on ice to form an oil-in-water emulsion.
- Solvent Evaporation: Stir the emulsion overnight at room temperature to allow complete evaporation of the organic solvent.
- Purification: Purify the formed celastrol-loaded nanoparticles by centrifugation or dialysis to remove free drug and surfactant.
- Characterization: Determine the particle size, zeta potential, and drug loading efficiency using dynamic light scattering and HPLC.
- Cellular Treatment: Apply the nanoformulated celastrol to cells and proceed with downstream target identification assays (e.g., Chemical Proteomics, Protocol 2).
Protocol 2: Chemical Proteomics for Unbiased Target Fishing
Principle: This strategy uses a functionalized, cell-permeable derivative of celastrol as a molecular bait to pull down and identify its interacting protein partners directly from a complex cellular lysate [34].
Materials:
- Celastrol derivative with a clickable handle (e.g., alkyne-tagged celastrol)
- Cell lysate from a relevant tissue or cell line
- Azide-functionalized solid support (e.g., agarose beads)
- Copper(I) catalyst (for click chemistry)
- Lysis buffer (e.g., RIPA buffer with protease inhibitors)
- Mass spectrometry (MS)-grade water, acetonitrile, and trypsin
Methodology:
- Probe Incubation: Incubate the alkyne-tagged celastrol probe with the prepared cell lysate for several hours to allow target engagement.
- Click Chemistry Conjugation: Perform a copper-catalyzed azide-alkyne cycloaddition (CuAAC) "click" reaction to covalently link the drug-protein complexes to the azide-functionalized solid support.
- Pull-Down and Wash: Thoroughly wash the beads with lysis buffer and high-salt buffers to remove non-specifically bound proteins.
- Protein Elution and Digestion: Elute the bound proteins using SDS-PAGE loading buffer or by on-bead digestion with trypsin.
- Mass Spectrometry Analysis: Analyze the resulting peptides by liquid chromatography-tandem mass spectrometry (LC-MS/MS) to identify the proteins that directly interacted with the celastrol probe.
Protocol 3: Thermal Proteome Profiling (TPP)
Principle: This label-free method monitors protein thermal stability changes induced by drug binding across the entire proteome. A compound's engagement with its target often makes the protein more or less resistant to heat-induced denaturation [34].
Materials:
- Celastrol (native or nanoformulated)
- Control vehicle (e.g., DMSO)
- Cultured cells of interest
- Multi-well PCR plate and thermal cycler
- Lysis buffer (with detergent and protease inhibitors)
- Centrifugal filter units
- MS sample preparation kit and LC-MS/MS instrumentation
Methodology:
- Cell Treatment: Divide a cell suspension into two aliquots. Treat one with celastrol and the other with a vehicle control.
- Heat Denaturation: For each condition, aliquot the cells into 10 tubes and heat them at different temperatures (e.g., from 37°C to 67°C) in a thermal cycler.
- Cell Lysis and Soluble Protein Collection: Lyse the heated cells and separate the soluble (non-denatured) protein fraction by centrifugation.
- Protein Quantification: Quantify the soluble protein in each temperature fraction for both treated and control samples using a method like tryptophan fluorescence or a proteomic approach.
- Data Analysis: Calculate the melting curve for each protein. A significant shift in the melting temperature (Tm) between the celastrol-treated and control samples indicates a direct or indirect target interaction.
Research Reagent Solutions
The following table details key reagents and materials essential for conducting the described target identification experiments for impermeable compounds.
Table 1: Essential Research Reagents for Target Identification Studies
| Reagent/Material | Function/Application | Key Considerations |
|---|---|---|
| Alkyne-tagged Celastrol Probe | Serves as bait in chemical proteomics to pull down target proteins [34]. | The tag must be attached at a site that does not disrupt its biological activity. |
| Nanocarriers (e.g., PLGA, Liposomes) | Enhance celastrol's solubility and cellular delivery, enabling intracellular target engagement [36] [35]. | Biocompatibility, drug loading efficiency, and release kinetics are critical parameters to optimize. |
| Permeability Assays (PAMPA, Caco-2) | Provide initial, high-throughput assessment of a compound's inherent permeability [40] [17]. | Cell-free PAMPA is suited for early screening, while cell-based Caco-2 offers more physiological relevance. |
| Microphysiological Systems (Organs-on-Chip) | Advanced in vitro models that mimic human physiology for more predictive permeability and efficacy studies [39]. | Can incorporate primary human cells, air-liquid interfaces, and fluid flow. |
| Mass Spectrometry-Grade Trypsin | Digests purified proteins into peptides for identification by LC-MS/MS in proteomic workflows [34]. | High purity is required to avoid non-specific cleavage and background noise. |
| Fixation/Permeabilization Reagents (e.g., Formaldehyde, Methanol) | Prepare cells for intracellular staining and analysis by flow cytometry or microscopy [38]. | Optimization is required to balance preserving cell structure and allowing antibody/probe access. |
Signaling Pathways and Experimental Workflows
Diagram 1: Integrated Strategy for Target Identification of Impermeable Compounds
Diagram 2: Key Signaling Pathways Modulated by Celastrol
Data Presentation: Quantitative Analysis of Celastrol's Properties and Models
Table 2: Physicochemical and Pharmacokinetic Properties of Celastrol [36] [35]
| Parameter | Value | Context / Implication |
|---|---|---|
| Aqueous Solubility | 13.25 ± 0.83 μg/mL | Classified as poorly soluble, limiting absorption [35]. |
| Absolute Oral Bioavailability | 17.06% (Rat) | Low systemic exposure after oral administration [36]. |
| Cmax (Oral, Rat) | 66.93 ± 10.28 μg/L | Low peak plasma concentration [36]. |
| Tmax (Oral, Rat) | 6.05 ± 1.12 h | Slow absorption rate [36]. |
| Therapeutic Dose (Mouse models) | 1 - 5 mg/kg | Effective in various xenograft models [35]. |
| Toxic Dose (Mouse models) | ~3 mg/kg and above | Narrow therapeutic window with reported adverse events (weight loss, organ toxicity) [35]. |
Table 3: Comparison of Permeability Assessment Models [39] [40] [17]
| Model Type | Example | Advantages | Disadvantages |
|---|---|---|---|
| Cell-Free | PAMPA (Parallel Artificial Membrane Permeability Assay) | High-throughput, low-cost, good for early screening of passive permeability [40]. | Lacks biological complexity (transporters, metabolism) [40]. |
| Cell-Based (Monolayer) | Caco-2, MDCK | More physiologically relevant than PAMPA; can study active transport [17]. | Extended cultivation time (Caco-2); may not fully represent target tissue [17]. |
| Advanced Co-culture | Caco-2/HT29-MTX | Incorporates mucin production, better simulating the intestinal barrier [17]. | More complex culture protocol. |
| Microphysiological System (MPS) | Small Airway-on-a-Chip | Recapitulates 3D architecture, air-liquid interface, and dynamic flow; highly physiologically relevant [39]. | Higher cost, more specialized equipment and expertise required [39]. |
Technical Support Center
Troubleshooting Guide: Common Issues & Solutions
Question: My CRISPR-Cas9 system is not efficiently editing the target site. What could be wrong?
Low editing efficiency can result from several factors. First, verify your guide RNA (gRNA) design, ensuring it targets a unique genomic sequence and has optimal length [41]. Second, confirm your delivery method is effective for your specific cell type; different cells may require optimization of electroporation parameters or alternative approaches like lipofection or viral vectors [41]. Finally, check the expression levels of Cas9 and the gRNA. Inadequate expression can be addressed by using a promoter suitable for your cell type, performing codon optimization of the Cas9 gene, and ensuring the quality and concentration of your plasmid DNA or mRNA are high [41].
Question: How can I minimize off-target effects in my CRISPR screen?
To minimize off-target activity, design highly specific gRNAs. Utilize online algorithms that predict potential off-target sites to help you optimize the gRNA sequence [41]. Furthermore, consider employing high-fidelity Cas9 variants, which have been engineered to significantly reduce off-target cleavage [41].
Question: I am working with rare primary cell samples and have low cell numbers. Is high-throughput screening still feasible?
Yes, recent technological advances have made this possible. Next-generation digital microfluidics (DMF) electroporation platforms enable high-throughput, low-input genome engineering. These systems can perform high-efficiency transfections using as few as 3,000 primary human cells per condition, a 100-fold reduction compared to conventional cuvette-based systems [42]. This miniaturization allows for parallelized experiments with precious cell populations.
Question: Why do different sgRNAs targeting the same gene show variable performance in my screen?
In the CRISPR/Cas9 system, gene editing efficiency is highly influenced by the intrinsic properties of each sgRNA sequence. It is common for different sgRNAs targeting the same gene to exhibit substantial variability in editing efficiency. To enhance the reliability of your results, it is recommended to design at least 3–4 sgRNAs per gene. This strategy mitigates the impact of individual sgRNA performance variability [43].
Question: If no significant gene enrichment is observed in my screen, what should I do?
The absence of significant enrichment is often due to insufficient selection pressure during the screening process, rather than a statistical error. When selection pressure is too low, the experimental group may fail to exhibit the intended phenotype, weakening the signal-to-noise ratio. It is recommended to increase the selection pressure and/or extend the screening duration to allow for greater enrichment of positively selected cells [43].
Question: How can I determine whether my CRISPR screen was successful?
The most reliable method is to include well-validated positive-control genes and their corresponding sgRNAs in your library. If these controls are significantly enriched or depleted as expected, it strongly indicates effective screening conditions [43]. In the absence of known controls, you can evaluate performance by assessing the cellular response (e.g., degree of cell killing) and examining bioinformatics outputs like the distribution and log-fold change of sgRNA abundance [43].
Quantitative Performance of Miniaturized Platforms
The table below summarizes key quantitative data from a miniaturized Digital Microfluidics (DMF) platform compared to a conventional system [42].
Table 1: Performance Comparison of Electroporation Platforms
| Parameter | Conventional System (Lonza Nucleofector) | Miniaturized DMF Platform |
|---|---|---|
| Minimum Viable Cell Input (per edit) | 100,000 - 250,000 cells | 3,000 - 10,000 cells |
| Throughput | Limited | 48 independently programmable reactions |
| Transfection Efficiency (Primary Human T cells, EGFP mRNA) | ~2% at 10,000 cells/edit | >90% (at 10,000 cells/edit) |
| Transfection Efficiency (Primary Myoblasts, EGFP mRNA) | <10% at 10,000 cells/edit | ~77% (at 3,000 cells/edit) |
| Automation Compatibility | Poorly suited | Seamless integration with laboratory automation |
Experimental Protocols for Key Assays
Protocol 1: Miniaturized Permeability Assay on Microcarriers (MAP)
This protocol is designed for high-throughput, genome-wide screens to identify genes regulating endothelial monolayer permeability [44].
- Cell Seeding: Grow endothelial cell monolayers on ~150 μm microcarriers (MCs). Each MC functions as an individual miniature permeability assay.
- CRISPR Perturbation: Transduce cells with a CRISPR library (e.g., sgRNAs targeting known thrombin signaling proteins) to generate knockout populations.
- Stimulation: Treat the MCs with thrombin to induce disassembly of intercellular junctions and increase monolayer permeability.
- Sorting and Analysis: Use fluorescence-assisted cell sorting to separate MCs carrying cells that resist thrombin-induced permeability (e.g., those expressing effective sgRNAs) from control MCs.
- Hit Identification: Isolate genomic DNA from sorted populations and sequence the integrated sgRNAs to identify genes whose knockout blocked the thrombin effect.
Protocol 2: Direct Measurement of Intracellular Compound Concentration
This protocol uses RapidFire Mass Spectrometry for high-throughput measurement of intracellular drug concentrations, providing insights into cell permeability and target engagement [9].
- Cell Preparation: Incubate HeLa cells (or other relevant cell types) with the test compound.
- Sample Processing: Rapidly process cells to isolate the intracellular fraction.
- Mass Spectrometry Analysis: Use a RapidFire-MS system for high-throughput, label-free quantification of the unbound drug concentration inside the cells.
- Data Calculation: Determine the intracellular bioavailability (Fic), which is the fraction of the extracellularly added compound that is bioavailable inside the cell. This platform can profile up to 100 compounds per day [9].
Signaling Pathways and Experimental Workflows
The following diagram illustrates the workflow for a miniaturized digital microfluidics (DMF) platform used in CRISPR screening.
The workflow for determining intracellular drug bioavailability (Fic) to bridge biochemical and cellular assay results is shown below.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for CRISPR-Compatible Miniaturized Assays
| Item | Function/Description | Example Application |
|---|---|---|
| Digital Microfluidics (DMF) Electroporation Cartridge | Planar electrode array for manipulating discrete droplets; enables high-throughput, low-input electroporation [42]. | Miniaturized genome engineering in primary cells (e.g., T cells, myoblasts). |
| sgRNA Library | A pooled or arrayed collection of single guide RNAs targeting genes across the genome. | Functional genomics screens to identify gene regulators of a phenotype. |
| High-Fidelity Cas9 Nuclease | An engineered Cas9 variant that reduces off-target cleavage while maintaining on-target activity [41]. | Improving the specificity of genetic edits in CRISPR screens. |
| Fluorescein Diacetate (FDA) | A lipophilic, non-fluorescent dye that becomes hydrophilic and fluorescent upon cleavage by intracellular esterases [33]. | Assessing cell membrane integrity and permeability in assay development. |
| Microcarriers (MCs) | ~150 μm beads for growing cell monolayers, functioning as individual miniature permeability assays [44]. | High-throughput genome-wide screens for genes regulating endothelial barrier function. |
Solving Permeability Challenges: Practical Strategies for Enhanced Cellular Uptake
Technical Specifications & Quantitative Data
The superior intracellular bioavailability of pterostilbene over resveratrol is grounded in key physicochemical and biological parameters. The data below quantifies these differences for easy comparison and experimental planning.
Table 1: Comparative Physicochemical Properties of Resveratrol and Pterostilbene
| Parameter | Resveratrol | Pterostilbene | Experimental/Methodological Context |
|---|---|---|---|
| Chemical Structure | 3 hydroxyl (-OH) groups [45] | 2 methoxy (-OCH₃) groups, 1 hydroxyl (-OH) group [45] | Core 1,2-diphenylethylene structure with different ring substitutions [45] [46]. |
| Lipophilicity (LogD) | Lower | Higher [47] | Determined via the shake-flask method; higher LogD indicates greater affinity for lipid environments [47]. |
| Cellular Uptake | Lower intracellular accumulation [47] | ~2.3x higher intracellular accumulation (in one enantiomer study) [1] | Measured via fluorescence microscopy and flow cytometry using cyanine2-labeled compounds in IPEC-J2 cells and porcine myotubes [47]. |
| Intracellular Bioavailability (Fic) | Lower | Higher [1] | Fic represents the fraction of unbound, bioactive drug inside the cell; measured using a label-free method [1]. |
| Metabolic Stability | Lower (susceptible to glucuronidation/sulfation) [47] | Higher (methoxy groups resist rapid metabolism) [47] | Based on the susceptibility of hydroxyl vs. methoxy groups to phase II metabolic enzymes [47]. |
Table 2: Functional Biological Outcomes in Photoaging and Antioxidant Assays
| Biological Activity | Resveratrol Performance | Pterostilbene Performance | Experimental Model & Key Findings |
|---|---|---|---|
| Anti-Photoaging | Significantly improves skin photoaging [48] | Preliminary evidence suggests potential to outperform resveratrol [48] | Based on a scoping review of in vitro, in vivo, and human trials; more research is needed for pterostilbene [48]. |
| Intracellular Antioxidant Capacity | Weaker intracellular ROS-scavenging capacity [47] | Stronger intracellular ROS-scavenging capacity [47] | Assessed in IPEC-J2 cells and porcine myotubes; directly linked to its higher cellular accumulation [47]. |
| General Pharmacological Properties | Potent anti-inflammatory, anti-cancer, neuroprotective effects [45] | Often exhibits stronger activity in comparable studies [45] [49] | The enhanced lipophilicity and bioavailability of pterostilbene are credited for its stronger potency in various disease models [45] [49]. |
Troubleshooting Guide & FAQs
This section addresses common challenges in intracellular target and permeability assays, with specific guidance derived from the resveratrol-pterostilbene comparative model.
Frequently Asked Questions (FAQs)
FAQ 1: Why does my compound show excellent biochemical potency but fails in cellular assays? This is a classic "cell drop-off" phenomenon, often due to inadequate intracellular bioavailability (Fic) [1].
- Solution: Measure the intracellular bioavailability (Fic) of your compound. A low Fic indicates that the compound is not effectively reaching its intracellular target, which can be due to poor membrane permeability, active efflux, or extensive intracellular binding [1]. Do not rely solely on artificial membrane permeability assays (like PAMPA), as they may not correlate well with actual cellular uptake [1].
FAQ 2: How can I experimentally verify and compare the cell membrane permeability of two analogous compounds? The comparative study on resveratrol and pterostilbene provides a robust methodological blueprint.
- Solution:
- Predict Permeability: Use Molecular Dynamics (MD) simulations, specifically the Potential of Mean Force (PMF) method, to predict and compare the membrane penetration capabilities of the compounds [47].
- Measure Uptake: Label the compounds with a fluorescent tag (e.g., Cyanine2) and treat relevant cell lines. Use fluorescence microscopy for visual confirmation of localization and flow cytometry for quantitative comparison of intracellular accumulation [47].
- Correlate with Function: Link the differences in uptake to a functional downstream outcome, such as the capacity to scavenge intracellular ROS [47].
FAQ 3: I am getting weak or no signal in my intracellular staining flow cytometry experiment. What could be wrong? Weak signal can stem from multiple sources in the experimental workflow.
- Solution:
- Permeabilization: Ensure adequate fixation and permeabilization. Use ice-cold methanol added drop-wise while vortexing, or a validated detergent like Triton X-100 [50] [51].
- Antibody/Fluorochrome: The detection antibody may be at too low a concentration. Furthermore, large fluorochromes can have difficulty entering cells; try a smaller, brighter dye [50] [51].
- Controls: Always include appropriate controls (unstained, isotype, positive control) to validate your protocol [50].
FAQ 4: How do I reduce high background in flow cytometry? High background is often caused by non-specific binding or the presence of dead cells.
- Solution:
- Titrate Antibodies: Use the optimal concentration of antibody to minimize non-specific binding [51].
- Blocking: Use Fc receptor blocking reagents or serum to block non-specific sites [50] [51].
- Viability Staining: Use a viability dye (e.g., PI, 7-AAD) to gate out dead cells, which exhibit high autofluorescence and non-specific binding [50] [51].
- Washing: Perform additional wash steps after antibody incubations to remove unbound reagent [51].
Troubleshooting Table: Intracellular Assay Challenges
| Problem | Possible Cause | Recommended Solution |
|---|---|---|
| High background in flow cytometry | Presence of dead cells [51] | Use a viability dye (e.g., PI, 7-AAD) and gate out dead cells during analysis [50] [51]. |
| Non-specific antibody binding [50] | Block with BSA, Fc receptor blockers, or normal serum. Include an isotype control [50] [51]. | |
| Weak fluorescence signal in flow cytometry | Inadequate cell permeabilization [50] [51] | Optimize permeabilization protocol (e.g., ice-cold methanol). Ensure reagents are fresh [50]. |
| Low expression of target protein [51] | Use a brighter fluorochrome (e.g., PE) for low-density targets [50]. Incorporate a positive control [51]. | |
| Compound shows cellular potency loss ("drop-off") | Low intracellular bioavailability (Fic) [1] | Measure Fic directly. Consider structural modifications to improve permeability and avoid efflux [1]. |
| Efflux by transporter proteins [1] | Test uptake in the presence of a pan-inhibitor like cyclosporine A to identify active efflux [1]. |
Research Reagent Solutions
The following reagents and tools are essential for conducting research inspired by the resveratrol-pterostilbene paradigm.
Table 3: Essential Research Reagents and Their Applications
| Reagent / Tool | Function / Principle | Application in Permeability Studies |
|---|---|---|
| Cyanine2 (CY2) Dye | A fluorescent label for tracking molecules [47]. | Chemically conjugate to compounds of interest (e.g., RES/PTS) to visualize and quantify their cellular uptake using microscopy and flow cytometry [47]. |
| Propidium Iodide (PI) / RNase Staining Solution | PI intercalates into double-stranded DNA; RNase ensures RNA is digested for DNA-specific staining [50]. | Used in cell cycle analysis by flow cytometry to resolve G0/G1, S, and G2/M phases. Also used as a viability dye to gate out dead cells [50]. |
| Saponin / Triton X-100 | Detergents that create pores in lipid membranes [50]. | Critical for permeabilizing cell membranes after fixation in intracellular staining protocols, allowing antibodies to access internal targets [50] [51]. |
| Cyclosporine A | A pan-inhibitor of active transport processes, including efflux pumps [1]. | Used in Fic assays to investigate whether a compound is a substrate for active efflux transporters, which would limit its intracellular accumulation [1]. |
| Brefeldin A | A Golgi transport blocker that inhibits protein secretion [51]. | Used in intracellular cytokine staining to cause proteins to accumulate within the cell, enhancing detection signal [51]. |
| Fixable Viability Dyes | Dyes that covalently bind to amines in dead cells and withstand fixation [50]. | Essential for distinguishing live from dead cells in fixed samples for flow cytometry, improving data accuracy by gating out dead cells that cause high background [50]. |
Conceptual Diagrams & Workflows
The following diagrams, generated using DOT language, illustrate the core concepts and experimental pathways for analyzing compound permeability.
Diagram 1: Stilbene Structure-Activity Relationship
Diagram 2: Intracellular Bioavailability Workflow
Troubleshooting Guide: Common P-gp Experimental Challenges
Problem: Inconsistent cellular accumulation results in permeability assays
- Potential Cause: Variable P-gp expression levels between cell passages or batches.
- Solution: Regularly validate P-gp expression and function in your cell lines using a reference substrate (e.g., digoxin, fexofenadine) and inhibitor (e.g., verapamil, zosuquidar). Use low-passage cells and maintain consistent culture conditions [1].
Problem: Lead compound shows high biochemical potency but low cellular activity
- Potential Cause: The compound is a P-gp substrate and is being actively effluxed from the cells, reducing its intracellular bioavailability (Fic).
- Solution: Determine the intracellular bioavailability (Fic) using a method that measures the unbound drug concentration inside the cell. Compare the cellular IC50 with the biochemical IC50; a significant drop ("cell drop-off") suggests efflux. Co-incubate with a P-gp inhibitor to see if cellular potency is restored [1].
Problem: Unexpectedly high toxicity in animal studies or clinical trials when a drug is co-administered with another
- Potential Cause: Pharmacokinetic drug-drug interaction where the co-administered drug inhibits P-gp, leading to increased absorption and reduced clearance of the investigational drug.
- Solution: Conduct thorough in vitro transport assays early in development to identify if your drug is a P-gp substrate. During in vivo studies, closely monitor plasma levels, especially if the drug has a narrow therapeutic index [52] [53].
Problem: Failure to reverse Multi-Drug Resistance (MDR) in cancer cell lines using a P-gp inhibitor
- Potential Cause: Redundancy in efflux transporters (e.g., overexpression of MRP1 or BCRP in addition to P-gp) or the involvement of non-transporter related resistance mechanisms.
- Solution: Characterize the full efflux transporter profile of the resistant cell line. Use broad-spectrum inhibitors or a combination of specific inhibitors. Consider using nanocarriers that can bypass multiple efflux mechanisms [54] [55].
Frequently Asked Questions (FAQs)
Q1: What is the functional consequence of P-gp overexpression in cancer cells? A: P-gp acts as a broad-spectrum efflux pump at the cell membrane. Its overexpression actively pumps out a wide range of structurally diverse chemotherapeutic agents, reducing intracellular drug accumulation to sub-therapeutic levels. This leads to multidrug resistance (MDR), a major obstacle in successful cancer chemotherapy [56] [55].
Q2: My compound is not a CYP3A4 substrate/inhibitor. Do I still need to test for P-gp interaction? A: Yes. Although P-gp and CYP3A4 have overlapping substrate specificities, their interactions are not always linked. A compound can be a P-gp substrate without significantly affecting or being affected by CYP3A4. Regulatory guidelines recommend dedicated tests for P-gp interactions [53].
Q3: What is the difference between inhibiting and bypassing P-gp? A: Inhibition uses a pharmacological agent (e.g., verapamil, cyclosporine A) to directly block the transporter's activity, preventing it from ejecting the co-administered drug. Bypassing uses formulation strategies, such as encapsulating the drug in liposomes or nanoparticles, which allows the drug to enter the cell via endocytosis, effectively evading recognition by P-gp [54] [52].
Q4: Are there any clinical successes in overcoming P-gp mediated MDR? A: Clinical results have been mixed. While many trials adding P-gp inhibitors to chemotherapy for solid tumors have been disappointing, some successes have been seen in hematologic malignancies. For example, a study in acute myeloid leukemia (AML) showed that adding cyclosporine A to daunorubicin improved relapse-free and overall survival by inhibiting P-gp and increasing intracellular daunorubicin concentrations [52].
Q5: How does the "floppase model" explain P-gp's mechanism? A: The floppase model proposes that P-gp captures its substrates from the inner leaflet of the lipid bilayer, not from the aqueous cytoplasm. Amphiphilic drugs partition into the membrane, where P-gp binds them and "flops" them to the outer leaflet, from which they can diffuse into the extracellular space. This model is supported by the fact that drug concentrations in the lipid phase are orders of magnitude higher than in the aqueous phase [55].
The following table classifies the effect of various P-gp modulators based on in vivo clinical studies using probe substrates like digoxin, dabigatran, and fexofenadine. The classification is adapted from FDA criteria, using the Area Under the Curve Ratio (AUCR) to define interaction strength [53].
| Interaction Strength | AUCR (Inhibitor) | AUCR (Inducer) | Representative Examples |
|---|---|---|---|
| Potent | ≥ 5 | ≤ 0.2 | Very few classified as potent (e.g., certain cyclosporine A regimens) [53]. |
| Moderate | ≥ 2 to < 5 | > 0.2 to ≤ 0.5 | Verapamil, Quinidine, Cyclosporine A [52] [53]. |
| Weak | ≥ 1.25 to < 2 | > 0.5 to < 0.8 | Clarithromycin, Itraconazole [53]. |
| Non-Interactor | < 1.25 | ≥ 0.8 | Levothyroxine, many others [53]. |
Note: The potential for a clinically significant interaction increases dramatically when a modulator affects both P-gp and CYP3A4, as a large proportion of P-gp modulators do [53].
Experimental Protocols for Assessing P-gp Interactions
Protocol 1: Determining Intracellular Bioavailability (Fic)
This label-free method measures the fraction of unbound, bioactive drug inside the cell, which is critical for engaging intracellular targets [1].
- Cell Preparation: Use a relevant cell type (e.g., PBMCs, Caco-2, or disease-specific cell lines). Grow cells to the desired confluence.
- Compound Incubation: Expose the cells to the test compound at a chosen concentration for a specified time.
- Measurement of Cellular Accumulation (Kp):
- After incubation, wash the cells thoroughly with a cold buffer to remove extracellular compound.
- Lyse the cells and measure the total drug concentration using a sensitive method like LC-MS/MS.
- Normalize the concentration to the cell volume or protein content to obtain Kp (the cell-to-medium concentration ratio).
- Measurement of Unbound Fraction (fu,cell):
- Use a method like equilibrium dialysis of the cell homogenate or a similar technique to determine the fraction of drug that is not bound to cellular components.
- Calculation:
- Calculate Fic using the formula: Fic = fu,cell × Kp.
- A low Fic value indicates poor intracellular exposure, often due to efflux transporters like P-gp [1].
Protocol 2: In Vitro P-gp Substrate Identification Assay
This assay determines if a drug is a substrate of P-gp by comparing its transport in two directions across a polarized cell monolayer.
- Cell Model: Use a polarized cell monolayer with high P-gp expression, such as Caco-2, MDCK-MDR1, or LLC-PK1-MDR1.
- Bidirectional Transport:
- A-to-B (Apical-to-Basolateral): Add the test compound to the apical side and measure its appearance in the basolateral chamber over time.
- B-to-A (Basolateral-to-Apical): Add the test compound to the basolateral side and measure its appearance in the apical chamber.
- With and Without Inhibitor: Perform both A-to-B and B-to-A transport studies in the presence and absence of a specific P-gp inhibitor (e.g., 10 µM cyclosporine A or 1 µM zosuquidar).
- Data Analysis:
- Calculate the apparent permeability (Papp) for each direction.
- Determine the efflux ratio: ER = Papp (B-to-A) / Papp (A-to-B).
- Interpretation: A compound is considered a P-gp substrate if the ER is high (e.g., > 2) and this ratio is significantly reduced (e.g., by ≥ 50%) in the presence of a P-gp inhibitor [56] [53].
Visualization of P-gp Mechanism and Experimental Workflow
P-gp Floppase Mechanism and Inhibition
Workflow for Identifying P-gp-Related Cell Drop-Off
The Scientist's Toolkit: Key Research Reagents & Materials
| Reagent / Material | Function / Application | Key Details & Examples |
|---|---|---|
| Cell Lines | In vitro models for transport and resistance studies. | Caco-2: Human colorectal adenocarcinoma, expresses P-gp. MDCK-MDR1: Madin-Darby canine kidney cells transfected with human MDR1 gene. Resistant cancer lines: Selected for MDR by chronic drug exposure [56] [1]. |
| Reference Substrates | Probe compounds to validate P-gp activity in assays. | Digoxin: Classic cardiac glycoside; FDA-recommended renal P-gp probe. Fexofenadine: Antihistamine; FDA-recommended intestinal P-gp probe. Dabigatran etexilate: Oral anticoagulant prodrug; P-gp probe [53]. |
| Reference Inhibitors | Pharmacological tools to confirm P-gp-specific effects. | Verapamil: First-generation inhibitor (calcium channel blocker). Cyclosporine A / Valspodar (PSC833): Potent second-generation inhibitor. Zosuquidar: Third-generation, highly specific P-gp inhibitor [55] [52]. |
| Nanocarrier Systems | Formulation approach to bypass P-gp recognition and efflux. | Polymeric Nanoparticles: e.g., PLGA-based. Liposomes: Lipid bilayer vesicles. Mixed Micelles: e.g., using Soluplus and Pluronic P105. Function: Protect payload, promote endocytic uptake [54]. |
| ATP Depletion Agents | Tool to study ATP-dependence of efflux. | Sodium Azide: Inhibits cellular respiration. 2-Deoxy-D-glucose: Glycolysis inhibitor. Used to confirm that P-gp-mediated efflux is an energy-dependent process [57]. |
Achieving efficient intracellular delivery of therapeutic agents is a central challenge in modern drug development. Biological membranes pose a significant barrier to the cellular uptake of many drugs, particularly large molecules and those with poor permeability characteristics. Advanced delivery systems, including liposomes, nanoparticles, and sophisticated conjugation technologies, have emerged as powerful tools to overcome these hurdles. These systems enhance cellular permeability, protect therapeutic cargo from degradation, and facilitate targeted delivery to intracellular sites of action. This technical support center provides troubleshooting guidance and foundational protocols for researchers developing these advanced delivery systems for intracellular target assays.
Troubleshooting Guide: Liposome and Nanoparticle Development
FAQ 1: How can I improve the encapsulation efficiency of hydrophilic drugs in liposomes?
Problem: Low encapsulation efficiency for hydrophilic (water-soluble) drugs leads to wasted materials and reduced therapeutic potential.
Solutions:
- Implement Active Loading Techniques: Establish a pH gradient (e.g., ammonium sulfate gradient) or ion gradient across the liposome membrane after formation. This "remote loading" technique can dramatically increase encapsulation efficiency for certain drugs, such as doxorubicin, to levels above 90% [58].
- Switch Preparation Methods: Use the Reverse-Phase Evaporation (REV) method, which is known for achieving higher encapsulation efficiencies for hydrophilic drugs compared to standard thin-film hydration [58] [59].
- Optimize Lipid Composition: Incorporate cholesterol into the lipid bilayer to reduce membrane permeability and prevent drug leakage. The use of saturated phospholipids with higher phase transition temperatures can also enhance bilayer stability [58] [60].
FAQ 2: My nanoparticle formulation is unstable and aggregates. What can I do?
Problem: Nanoparticles aggregate in solution, leading to inconsistent results and potential experimental failure.
Solutions:
- Modify Surface Charge: Increase the absolute value of the zeta potential to greater than ±30 mV by incorporating charged lipids (e.g., cationic DOTAP or anionic DSPG). This enhances electrostatic repulsion between particles, improving colloidal stability [61].
- Introduce Steric Stabilizers: Incorporate PEGylated lipids (e.g., DSPE-PEG) at 1-5 mol% of the total lipid composition. The PEG polymer creates a steric "cloud" that prevents nanoparticles from coming close enough to aggregate [60] [61].
- Control Storage Conditions: Store formulations in sugar-based cryoprotectant solutions (e.g., 10% trehalose or sucrose) and consider lyophilization for long-term storage to prevent fusion and aggregation [62].
FAQ 3: How can I enhance the cellular uptake of my nanocarriers?
Problem: Despite successful formulation, cellular internalization of the nanocarrier is inefficient.
Solutions:
- Conjugate Cell-Penetrating Peptides (CPPs): Covalently attach CPPs like TAT (from HIV) or poly-arginine (R8/R9) to the nanoparticle surface. These peptides facilitate energy-independent direct penetration or energy-dependent endocytotic processes, greatly enhancing cellular uptake [63].
- Implement Active Targeting: Decorate the surface with targeting ligands (e.g., folate, transferrin, or monoclonal antibody fragments) that bind to receptors overexpressed on your target cells. This leverages receptor-mediated endocytosis for specific and efficient internalization [60] [62].
- Optimize Physicochemical Properties: Fine-tune the nanoparticle size. For most cell types, a size range of 80-150 nm is optimal for cellular uptake. A slightly positive surface charge (zeta potential) can also promote interaction with the negatively charged cell membrane [61] [63].
Table 1: Critical Quality Attributes (CQAs) for Lipid-Based Nanoparticles
| Parameter | Target Range | Impact on Performance | Characterization Technique |
|---|---|---|---|
| Particle Size | 50-200 nm (tailor to target cell) | Influences circulation time, tissue penetration, and cellular uptake [61] | DLS, NTA, TRPS [61] |
| Polydispersity Index (PDI) | < 0.2 (ideal), < 0.3 (acceptable) | Indicates population homogeneity; high PDI leads to inconsistent behavior [61] | DLS, MADLS [61] |
| Zeta Potential | > ±30 mV for stability; tune for uptake | Predicts colloidal stability; charge affects cell membrane interaction [61] [63] | Electrophoretic Light Scattering |
| Encapsulation Efficiency | > 80% (ideal) | Directly impacts drug loading capacity and cost-effectiveness [58] | HPLC/UHPLC after separation |
| Lamellarity | Unilamellar vs. Multilamellar | Affirms structural consistency and impacts drug release kinetics [59] | Cryo-TEM [61] |
Core Experimental Protocols
Protocol 1: Microfluidic Synthesis of Liposomes for High Reproducibility
Objective: To produce monodisperse, unilamellar liposomes with high batch-to-batch reproducibility using a microfluidic mixer.
Materials:
- Lipids (e.g., DSPC, Cholesterol, DSPE-PEG2000)
- Anhydrous ethanol and Phosphate Buffered Saline (PBS, pH 7.4)
- Syringe pumps (2)
- Microfluidic mixer chip (e.g., staggered herringbone or T-junction)
- Syringes and tubing
Method:
- Prepare Lipid Solution: Dissolve lipids in ethanol at a total concentration of 10-20 mg/mL.
- Prepare Aqueous Phase: Use PBS or the hydration buffer of your choice.
- Set Up Flow Rates: Load the lipid and aqueous solutions into separate syringes on syringe pumps. A typical Total Flow Rate (TFR) is 10-15 mL/min with a Flow Rate Ratio (FRR, aqueous:organic) of 3:1. Higher FRR generally yields smaller particles [64].
- Mixing: Simultaneously pump both solutions through the microfluidic mixer. The rapid and homogeneous mixing triggers liposome self-assembly.
- Dialyze or Ultrafiltrate: Immediately process the output stream to remove residual ethanol and buffer-exchange into the final storage buffer.
- Characterize: Analyze the final formulation for size, PDI, and concentration using DLS/NTA [61] [64].
Troubleshooting:
- Problem: Liposomes are too large. Solution: Increase the FRR or the TFR.
- Problem: Solution appears cloudy or precipitates. Solution: Ensure lipids are fully dissolved in the organic phase and check for compatibility between solvent and buffer.
Protocol 2: Conjugation of Cell-Penetrating Peptides (CPPs) to Nanoparticles
Objective: To covalently attach a CPP (e.g., TAT) to the surface of pre-formed, PEGylated nanoparticles for enhanced cellular uptake.
Materials:
- Pre-formed nanoparticles with maleimide-functionalized PEG lipids (e.g., DSPE-PEG-Mal)
- CPP with a terminal cysteine residue (e.g., Cys-TAT)
- Reducing agent (e.g., TCEP)
- Purification equipment (e.g., dialysis membrane, size exclusion chromatography)
Method:
- Activate Nanoparticles: Incubate nanoparticles with a mild reducing agent like TCEP to ensure the maleimide groups are reactive.
- Purify Nanoparticles: Remove excess TCEP via dialysis or gel filtration.
- Conjugate CPP: Add the cysteine-containing CPP to the nanoparticle suspension at a molar ratio of 1.5:1 (CPP:available maleimide). React for 4-6 hours at room temperature under gentle agitation.
- Purify Conjugate: Remove unreacted CPP by extensive dialysis or size exclusion chromatography.
- Verify Conjugation: Confirm successful conjugation by measuring an increase in particle size via DLS and a change in zeta potential. Use methods like HPLC or colorimetric assays to quantify unconjugated peptide in the filtrate [63].
Troubleshooting:
- Problem: Low conjugation efficiency. Solution: Ensure the CPP sequence has a single, accessible cysteine; avoid amine-containing buffers that can compete with the reaction.
- Problem: Nanoparticle aggregation post-conjugation. Solution: The CPP may be causing bridging; try a lower molar ratio of CPP to nanoparticle or introduce a spacer arm between the peptide and the particle surface.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents for Advanced Delivery System Research
| Reagent / Material | Function / Application | Key Considerations |
|---|---|---|
| DSPE-PEG | Imparts "stealth" properties, reduces opsonization, and improves circulation half-life [60]. | PEG chain length (e.g., PEG2000) and density (1-5 mol%) impact stealth effect and targeting [60]. |
| Ionizable/Cationic Lipids | Encapsulates nucleic acids (mRNA, siRNA) via electrostatic interaction; key component of LNPs [61]. | Ionizable lipids (e.g., DLin-MC3-DMA) are preferred for reduced toxicity compared to permanently cationic lipids. |
| Cholesterol | A "helper lipid" that fills gaps in the bilayer, enhancing membrane stability and rigidity [61] [59]. | Typically comprises 30-40 mol% of the lipid composition. |
| Cell-Penetrating Peptides (CPPs) | Enhances cellular internalization of nanoparticles via various translocation mechanisms [63]. | Cationic types (TAT, R8) are common; can be conjugated post-formation to avoid interference with self-assembly. |
| Targeting Ligands | Enables active targeting to specific cell types (e.g., folate, transferrin, monoclonal antibodies) [62]. | Requires post-insertion techniques or conjugation to terminal PEG groups to maintain functionality. |
| Microfluidic Mixer Chips | Enables reproducible, scalable production of nanoparticles with low PDI [64]. | Staggered herringbone mixers (SHM) provide efficient mixing for uniform self-assembly. |
Schematic Workflows
Liposome Preparation and Cellular Uptake Pathway
Liposome Development and Cellular Journey - This diagram outlines the workflow from key laboratory preparation methods through industrial scale-up, critical characterization steps, and the primary pathways for cellular uptake and intracellular delivery.
Strategies to Overcome Biological Barriers
Barriers and Engineering Solutions - This diagram maps specific biological delivery barriers against the corresponding nanoparticle engineering strategies designed to overcome them, providing a clear problem-solution framework for researchers.
FAQs on Permeability Assay Challenges
What are the primary sources of variability in cell-based permeability assays? Variability primarily stems from inconsistencies in cell monolayer integrity, sample processing, and critical reagent handling. Key factors include the quality and differentiation of the cell monolayer (e.g., Caco-2), the stability of test compounds, and the precision of analytical methods. Standardizing protocols for cell culture, compound handling, and instrument calibration is crucial for minimizing this variability [65] [66].
How can I improve the reproducibility of my intracellular staining for flow cytometry? Reproducibility hinges on optimized fixation and permeabilization steps. Use fresh, high-quality reagents and validate your protocol for your specific cell type and target. For instance, when using methanol for permeabilization, it is critical to chill cells on ice prior to drop-wise addition of ice-cold methanol to prevent hypotonic shock [67]. Furthermore, standardize antibody concentrations and incubation times, and always include appropriate controls like unstained, isotype, and positive controls to account for non-specific binding and instrument setup [67] [51].
My permeability data is inconsistent day-to-day. What should I check first? First, verify the integrity of your cell monolayers before each assay. For Caco-2 models, confirm that the Transepithelial Electrical Resistance (TEER) values meet pre-defined acceptance criteria (e.g., > 1000 Ω·cm² for 24-well plates) [68]. Second, ensure the consistency of critical reagents, such as using fresh serum lots and validating new batches of buffers. Third, implement a robust quality control process for your analytical instrumentation [65].
Troubleshooting Guides
Common Issues in Cell Monolayer Permeability Assays
| Problem | Possible Causes | Recommendations |
|---|---|---|
| Poor Monolayer Integrity | Inconsistent cell culture conditions; insufficient differentiation time; microbial contamination. | Use standardized, ready-to-use cell models where possible [68]; Validate monolayer integrity before each assay (e.g., TEER > 1000 Ω·cm², Lucifer Yellow Papp ≤ 1 x 10⁻⁶ cm/s) [68]. |
| High Variability in Papp Values | Inconsistent compound solubility/preparation; hydrolysis or binding of compound to plasticware; analytical error. | Include high/low permeability controls in every run (e.g., Propranolol and Atenolol) [68]; Perform assays in triplicate; Use mass spectrometry for sensitive and specific concentration analysis [66] [68]. |
| Inaccurate Efflux Ratio | Variable expression of efflux transporters like P-gp; over-differentiated cell monolayers. | Use well-characterized cell models (e.g., MDCK-MDR1) [69]; For Caco-2, consider a tiered approach using accelerated (5-day) and conventional (21-day) models for a definitive answer on P-gp substrates [66]. |
Common Issues in Intracellular Staining & Flow Cytometry
| Problem | Possible Causes | Recommendations |
|---|---|---|
| Weak or No Signal | Inadequate cell permeabilization; low target expression; suboptimal antibody concentration. | Optimize permeabilization conditions (e.g., agent concentration, duration) [70] [67]; Validate antigen expression with a known positive control; Titrate antibodies to find the optimal concentration [51]. |
| High Background Staining | Non-specific antibody binding; incomplete washing; presence of dead cells; over-fixation. | Block Fc receptors prior to staining [67]; Include additional wash steps with detergents like Tween or Triton in the buffer [51]; Use a viability dye to gate out dead cells [67]. |
| High Day-to-Day Variability | Inconsistent sample preparation; instrument setting drift; reagent lot changes. | Implement daily instrument quality control using calibrated beads [65] [71]; Use standardized antibody panels and clones across experiments [71]; Control sample processing variables like anticoagulant type and storage temperature rigorously [65]. |
The Scientist's Toolkit: Key Research Reagent Solutions
| Item | Function & Application |
|---|---|
| Saponin | A commonly used permeabilization agent that creates pores in the plasma membrane by complexing with cholesterol, allowing antibodies to access intracellular targets. It is often used in conjunction with formaldehyde fixation [67]. |
| Digitonin | A permeabilizing agent used in mitochondrial function assays and other applications. It is critical to optimize its concentration to make the plasma membrane permeable while keeping intracellular organelles intact [70]. |
| Recombinant Perfringolysin O | A recombinant bacterial toxin that creates large pores in cholesterol-containing membranes, used as an alternative permeabilization agent in functional assays [70]. |
| Ice-Cold Methanol | An effective permeabilizing and fixing agent, particularly for nuclear targets and cell cycle analysis. It is critical to add it drop-wise to a cell pellet while gently vortexing to prevent cell damage from hypotonic shock [67]. |
| Triton X-100 | A non-ionic detergent used for permeabilizing cell membranes. It is a common component in wash buffers to reduce background, but fresh solutions should be used for consistent results [67]. |
| MDCK-MDR1 Cells | Madin-Darby Canine Kidney cells overexpressing the human MDR1 (P-glycoprotein) transporter. This is a well-established model for evaluating whether a compound is a substrate or inhibitor of this key efflux transporter [69]. |
| Caco-2 Cells | A human colon carcinoma cell line that, upon differentiation, forms a polarized monolayer with brush border enzymes and transporters, making it the gold standard for predicting intestinal absorption [66] [68]. |
| Fixable Viability Dyes | These dyes (e.g., eFluor) covalently bind to amines in cells, allowing them to withstand fixation and permeabilization steps. They are essential for distinguishing live from dead cells during intracellular staining, thereby reducing background [67]. |
| Reference Compounds (Propranolol, Atenolol) | High and low permeability controls, respectively, used to validate the performance of Caco-2 and other permeability assays in every run [68]. |
Detailed Experimental Protocols
Optimized Protocol for 5-Day Cultured Caco-2 Permeability Assay
This accelerated model offers a high-throughput screen for permeability and P-gp efflux measurements [66].
Key Methodology:
- Cell Seeding and Culture: Plate Caco-2 cells (e.g., HTB-37) onto collagen-coated transwell inserts in a 24-well plate format. Culture the cells for 5 days in media containing sodium butyrate to accelerate differentiation and growth [66].
- Monolayer Integrity Check: Before the assay, verify monolayer integrity. Acceptable criteria include a TEER value > 1000 Ω·cm² for 24-well plates and a Lucifer Yellow apparent permeability (Papp) ≤ 1 x 10⁻⁶ cm/s [68].
- Permeability Assay:
- Analytical Detection: Quantify compound concentrations using a sensitive and specific method like liquid chromatography-tandem mass spectrometry (LC-MS/MS) [66].
- Data Analysis: Calculate the apparent permeability coefficient (Papp) using the formula:
- Papp = (dQ/dt) / (A × C0)
- Where dQ/dt is the permeation rate (nmol/s), A is the membrane surface area (cm²), and C0 is the initial donor concentration (nmol/mL) [68].
- Tiered Approach: Due to potentially lower P-gp expression, use the 5-day model for initial screening. Re-test compounds that are positive or borderline P-gp substrates in the conventional 21-day model for a definitive result [66].
Optimized Protocol for Intracellular Staining for Flow Cytometry
A robust protocol is essential for detecting intracellular proteins, such as cytokines, transcription factors, or signaling phospho-proteins.
Key Methodology:
- Cell Preparation and Surface Staining: Harvest and wash cells. First, stain for surface markers in buffer containing PBS and BSA. Use Fc receptor blocking reagents to minimize non-specific binding [67] [51].
- Fixation: Fix cells immediately after treatment to inhibit enzyme activity and preserve the cellular state. Use 4% methanol-free formaldehyde for a defined period (typically 10-15 minutes) to avoid epitope damage [67] [51].
- Permeabilization: Centrifuge cells and thoroughly resuspend the pellet in a permeabilization buffer. Common agents include saponin, Triton X-100, or ice-cold 90% methanol.
- Critical Step for Methanol: Chill cells on ice prior to drop-wise addition of ice-cold methanol while gently vortexing to ensure homogeneous permeabilization and avoid hypotonic shock [67].
- Intracellular Staining: Add fluorescently conjugated antibodies against the intracellular target to the permeabilized cells. Incubate in the dark for the optimized duration and temperature.
- Washing and Acquisition: Wash cells thoroughly with permeabilization buffer to remove unbound antibody, then resuspend in flow cytometry staining buffer. Acquire data on a flow cytometer, running samples at a low flow rate to improve resolution, especially for cell cycle analysis [67].
- Controls: Always include:
Experimental Workflow for Permeability and Intracellular Assays
The following diagrams outline the logical workflow for two key types of assays discussed, highlighting critical steps for minimizing variability.
Optimizing Cell-Based Permeability Assays
Optimizing Intracellular Staining for Flow Cytometry
Ensuring Data Reliability: Validation Frameworks and Cross-Assay Correlation
In the pursuit of overcoming cell permeability issues in intracellular target assays, robust Quantitative Structure-Property Relationship (QSPR) models serve as indispensable tools for predicting compound absorption. The accuracy of these models hinges entirely on the quality of the experimental data used to build them. The apparent permeability coefficient (Papp), derived from Caco-2 cell assays, is a critical parameter for estimating human intestinal permeability. Inconsistent Papp values introduce noise that severely compromises model reliability, leading to flawed predictions and misguided compound optimization. This technical support guide details the methodologies for generating consistent Papp data and troubleshooting common experimental variabilities that impede QSPR model development.
Experimental Protocols: Establishing a Reliable Caco-2 Assay
A standardized experimental protocol is the first and most crucial defense against data inconsistency.
Key Steps for a Successful Caco-2 Permeability Assay
The following workflow ensures the generation of high-quality, reproducible Papp values for QSPR modeling.
Caco-2 Cell Monolayer Preparation and Integrity Checks
- Culture Conditions: Seed Caco-2 cells on transwell inserts and culture for 15-21 days to allow full differentiation into an enterocyte-like monolayer [68]. Change the culture medium every two days [68].
- Integrity Validation: Before the assay, verify monolayer integrity using the following acceptance criteria [68]:
| Measurement | 24-Well Format | 96-Well Format |
|---|---|---|
| Transepithelial Electrical Resistance (TEER) | > 1000 Ω·cm² | > 500 Ω·cm² |
| Lucifer Yellow Papp (Paracellular Flux) | ≤ 1 x 10⁻⁶ cm/s | ≤ 1 x 10⁻⁶ cm/s |
Permeability Assay Execution
- Incubation Conditions: Perform the permeability assay for 2 hours. Test each compound in triplicate in both apical-to-basal (A-B) and basal-to-apical (B-A) directions. A starting compound concentration of 10 µM is recommended for unknown compounds [68].
- Reference Compounds: Always include high-permeability (e.g., Propranolol) and low-permeability (e.g., Atenolol) controls to validate the assay performance in each run [68].
Papp Calculation and Data Interpretation The apparent permeability coefficient (Papp) is calculated using the formula [68]:
Where:
- dQ/dt: Permeation rate (nmol/s)
- A: Surface area of the cell monolayer (cm²)
- C₀: Initial concentration of the compound in the donor compartment (nmol/mL)
The calculated Papp values can be correlated with predicted in vivo absorption using the following reference table [68]:
| In Vitro Papp Value | Predicted In Vivo Absorption |
|---|---|
| Papp ≤ 1.0 x 10⁻⁶ cm/s | Low (0-20%) |
| 1.0 x 10⁻⁶ cm/s < Papp ≤ 10 x 10⁻⁶ cm/s | Medium (20-70%) |
| Papp > 10 x 10⁻⁶ cm/s | High (70-100%) |
The Scientist's Toolkit: Essential Research Reagents
| Item | Function & Importance |
|---|---|
| Caco-2 Cell Line | A human colon adenocarcinoma cell line that, upon differentiation, mimics the intestinal epithelial barrier, forming tight junctions and expressing relevant transporters [17]. |
| Transwell Inserts | Semi-porous polyester filters that provide independent access to apical and basal compartments, enabling permeability measurements [68]. |
| Reference Compounds | High/Low permeability controls (e.g., Propranolol/Atenolol) and transporter substrates/inhibitors (e.g., Digoxin/Verapamil) for assay validation and system suitability testing [68]. |
| TEER Measurement System | An volt/ohmmeter used to non-invasively monitor the integrity and tight junction formation of the cell monolayer [68]. |
| Lucifer Yellow | A fluorescent paracellular marker used to quantify and ensure the integrity of the cellular monolayer by measuring its low passive diffusion [68]. |
| Sensitive Analytical Instrumentation | Liquid chromatography with tandem mass spectrometry (LC-MS/MS) is typically required for the specific and sensitive quantification of test compound concentrations in the receiver compartment [68]. |
Troubleshooting Guides & FAQs: Ensuring Data Consistency
Frequently Asked Questions
Q1: Our QSPR model performance is poor. Could inconsistent training data be the cause? Yes, this is a primary cause. Inconsistent Papp values from source data—often due to different experimental conditions across labs—introduce significant noise. For a reliable model, curate a large dataset (e.g., 1,800+ compounds) from a single source or rigorously standardize data from multiple sources by applying strict filters (e.g., removing Papp values < 10⁻⁸ or > 10⁻³.⁵ cm/s and averaging replicates) [73].
Q2: What are the most critical parameters to control for Papp assay reproducibility? The key parameters are:
- Cell Passage Number: Use cells within a narrow, low-passage range.
- Culture Duration: Maintain a consistent and sufficient differentiation period (15-21 days).
- Mono layer Integrity: Adhere strictly to TEER and Lucifer Yellow flux acceptance criteria before every assay.
- Assay Conditions: Standardize incubation time, temperature, pH, and buffer composition across all experiments [68].
Q3: How can we handle existing data from multiple laboratories for QSPR modeling?
- Data Curation: Critically evaluate and clean the data. Remove obvious outliers and compounds without clear structural identifiers (SMILES) [73].
- Standardization: Apply a uniform data-cleaning protocol. For compounds with multiple reported Papp values, take the arithmetic mean if the values are not significantly different. If values differ greatly, it is safer to exclude them from the modeling set [73].
- Define an Application Domain (AD): Use chemoinformatic methods to define the chemical space of your model. Predictions for compounds outside this AD should be treated with caution [73].
Advanced Data Analysis for Model Robustness
For researchers employing computational modeling, the choice of molecular features significantly impacts prediction accuracy, especially with high-quality Papp data.
Systematic benchmarking reveals that certain molecular feature representations yield superior performance for Caco-2 prediction [74]:
- Highly Effective Descriptors: PaDEL, Mordred, and RDKit descriptors are particularly effective, especially when 3D descriptors are included, which can reduce the Mean Absolute Error (MAE) by ~16% compared to using 2D features alone [74].
- Optimal Modeling Techniques: Automated Machine Learning (AutoML) frameworks like AutoGluon can efficiently search and ensemble top-performing algorithms (e.g., LightGBM, XGBoost), simplifying the model development process and often achieving state-of-the-art performance [75] [74].
- Feature Importance: Leverage tools like SHAP analysis to interpret the model and understand which molecular descriptors (e.g., related to hydrogen bonding or topological polar surface area) are most critical for permeability, providing a mechanistic check on the model's predictions [74].
Core Concepts and Troubleshooting
This guide addresses common challenges in correlating in vitro, in silico, and in vivo data, a critical step for successful drug development, particularly for compounds facing cell permeability issues with intracellular targets.
What are the different levels of In Vitro-In Vivo Correlation (IVIVC) and when is each level sufficient?
The U.S. Food and Drug Administration (FDA) defines several levels of IVIVC, which represent a predictive mathematical model between an in vitro property and a relevant in vivo response [76]. The choice of level depends on your development stage and regulatory goals.
The table below summarizes the key IVIVC levels:
| IVIVC Level | Description | Key Application / Regulatory Value |
|---|---|---|
| Level A | The most precise correlation; point-to-point relationship between the in vitro dissolution rate and the in vivo input rate (e.g., absorption rate) [76]. | Considered the gold standard; can be used as a surrogate for bioequivalence studies, supports waiver for post-approval changes [76]. |
| Level B | Uses statistical moment analysis; compares the mean in vitro dissolution time to the mean in vivo residence time or dissolution time [76]. | Less precise than Level A; often sufficient for internal formulation design and screening, but may not satisfy all regulatory requirements [76]. |
| Level C | Relates a single dissolution time point (e.g., t~50%~) to a single pharmacokinetic parameter (e.g., AUC or C~max~) [76]. | Useful for early development and product characterization; Multiple Level C (correlating several time points to PK parameters) provides more information and can be more valuable [76]. |
| Level D | A qualitative analysis rather than a formal correlation; serves as a rough rank-order comparison [76]. | No regulatory value; used only for initial formulation guidance and development [76]. |
For formulation design and screening, Level B and C correlations are often sufficient. However, for regulatory purposes like setting dissolution specifications or waiving bioequivalence studies, a Level A correlation is the target [76].
Why does my in vitro permeability data consistently overestimate the in vivo absorption for my lipid-based formulation?
This is a common issue, often stemming from the inability of simple in vitro assays to capture the complex dynamic processes that occur in vivo. The following troubleshooting guide addresses potential causes and solutions.
| Problem Area | Specific Issue | Recommended Troubleshooting Action |
|---|---|---|
| In Vitro Model Limitations | Using a basic dissolution test (e.g., USP apparatus) that does not account for lipid digestion [76]. | Transition to a more biorelevant in vitro lipolysis model. This assay incorporates digestive enzymes to simulate the processing of lipid-based formulations in the GI tract, providing a more accurate prediction of drug precipitation and solubilization [76]. |
| Using a single cell-line model (e.g., Caco-2) that lacks a mucosal layer [17]. | Improve physiological relevance by using a co-culture model, such as Caco-2 with HT29-MTX cells. The HT29-MTX cells produce mucin, creating a mucus layer that can more accurately represent a barrier to absorption [17]. | |
| Data Correlation & Modeling | Relying on a simple correlation model for a complex formulation [77]. | Employ a Physiologically Based Pharmacokinetic (PBPK) modeling approach. PBPK models integrate physicochemical drug properties, formulation data, and physiological parameters to better simulate and predict in vivo performance, especially for complex injectable drug products (CIDPs) and formulations exhibiting flip-flop pharmacokinetics [77]. |
| Failure to account for multiphasic drug release from a complex formulation [77]. | Ensure your in vitro release method is "discriminatory" and can capture multiple release phases (e.g., initial burst, sustained release). The model used for IVIVC must be customized to this complex release profile [77]. |
How can I improve the predictivity of my in silico permeability models?
Bridging the gap between in silico predictions and experimental results requires careful benchmarking and an understanding of the limitations of each method. Adhering to community-led guidelines is key [78].
- Benchmark Your Models Rigorously: Do not use your in silico tool in a vacuum. Systematically benchmark its predictions against reliable in vitro (e.g., Caco-2, PAMPA) and in vivo data. This process is sensitive to assay design choices, so the conditions of the experimental data used for benchmarking must be well-understood [78].
- Understand the "Black Box": Know the fundamental approach your in silico assay uses. The two primary methodologies are transition-based counting and the inhomogeneous-solubility diffusion approach. Understanding the basis of the calculation helps interpret results and identify potential failure points [78].
- Account for the Biological Barrier: A generic permeability model may not be suitable for all tissues. For targets like the central nervous system, use in silico models specifically tailored and validated for the blood-brain barrier (BBB), as its unique physiology demands specialized considerations [78].
- Validate with 3D and Advanced Models: As new in vitro models gain prominence, validate your in silico tools against them. Newer models like organ-on-a-chip systems and cell spheroids offer greater physiological relevance than traditional 2D cultures. A strong correlation with these systems increases confidence in your in silico predictions [17].
Essential Experimental Protocols
Protocol 1: In Vitro Lipolysis Assay for Lipid-Based Formulations
This protocol is critical for obtaining biorelevant dissolution data for Lipid-based Formulations (LBFs), which is essential for building a meaningful IVIVC [76].
- Simulated Gastrointestinal Media Preparation: Prepare simulated gastric and intestinal fluids. The intestinal medium typically contains taurocholate (bile salt) and phosphatidylcholine (lectithin) to mimic fed or fasted state conditions, as well as calcium ions to drive the precipitation of fatty acid soaps.
- pH-Stat Titration Setup: Place the lipolysis vessel in a thermostated water bath (typically 37°C). The pH of the medium is constantly monitored.
- Initial Gastric Phase (Optional): The formulation may be first introduced into simulated gastric fluid for a short period.
- Intestinal Lipolysis Phase: Transfer the content to the intestinal medium. Start the reaction by adding pancreatic lipase/colipase enzymes.
- Titration: As digestion occurs, fatty acids are released, which would lower the pH. A pH-Stat titrator automatically adds a sodium hydroxide (NaOH) solution to maintain a constant pH (typically 6.5). The volume of NaOH consumed over time is directly proportional to the extent of lipolysis.
- Sampling and Analysis: At predetermined time points, samples are withdrawn. The digestion process in these samples is often stopped to prevent further enzyme activity. Samples are then ultracentrifuged to separate into different phases (e.g., aqueous, pellet, oily). The drug concentration in each phase is quantified to understand its distribution and potential for precipitation.
Protocol 2: Parallel Artificial Membrane Permeability Assay (PAMPA)
PAMPA is a high-throughput, non-cell-based model used for early-stage passive permeability screening [17].
- Artificial Membrane Formation: A filter on a multi-well acceptor plate is coated with a lipid solution (e.g., lecithin in dodecane) to create an artificial membrane.
- Plate Assembly: The donor plate (containing the drug compound in buffer) is stacked on top of the acceptor plate, creating a sandwich where the artificial membrane separates the two compartments.
- Incubation: The assembled plate is incubated for several hours (e.g., 4-16 hours) at room temperature or 37°C without shaking to allow for passive diffusion.
- Disassembly and Quantification: After incubation, the plates are separated. The concentration of the drug in both the donor and acceptor compartments is measured using a method like UV spectroscopy or LC-MS/MS.
- Permeability Calculation: The apparent permeability coefficient (P~app~) is calculated based on the drug's appearance in the acceptor compartment and its disappearance from the donor compartment over time.
The Scientist's Toolkit: Key Research Reagent Solutions
The table below lists essential materials and their functions in permeability and correlation studies.
| Reagent / Material | Function in Experiment |
|---|---|
| Caco-2 Cell Line | A human colon adenocarcinoma cell line that, upon differentiation, forms a polarized monolayer with tight junctions and expresses brush border enzymes, simulating the human intestinal epithelium for permeability studies [17]. |
| MDCK Cell Line | Madin-Darby Canine Kidney cells; form tight monolayers with a shorter cultivation time than Caco-2 cells. Often used as a surrogate for passive permeability screening [17]. |
| HT29-MTX Cell Line | A mucin-producing human colon adenocarcinoma cell line. Used in co-culture with Caco-2 cells to introduce a physiologically relevant mucus layer to the permeability model [17]. |
| Pancreatic Lipase/Colipase | Digestive enzymes critical for the in vitro lipolysis assay. They hydrolyze triglycerides from lipid-based formulations, mimicking the dynamic solubilization and potential precipitation of a drug in the GI tract [76]. |
| Taurocholate & Phosphatidylcholine | Key components of simulated intestinal fluids, acting as bile salts and phospholipids to form micelles and colloidal structures that can solubilize lipophilic drugs [76]. |
| Electrospun Nanofiber Scaffolds | Synthetic scaffolds used to enhance the performance and more physiologically relevant 3D culture of Caco-2 and other cell lines, improving the model's predictability [17]. |
| PLGA/PLA Polymers | Poly(lactic-co-glycolic acid) and Poly(lactic acid); biodegradable polymers used to create complex injectable drug products (CIDPs) like microspheres. Their composition and molar mass modulate drug release kinetics [77]. |
Correlation Workflows and Data Relationships
Permeability Assay Selection Logic
Troubleshooting Guide: Common Experimental Issues
Problem: Inconsistent cellular potency results between biochemical and cellular assays.
| Problem Area | Possible Causes | Recommendations |
|---|---|---|
| Cell Drop-Off | Inadequate intracellular compound concentration; Low membrane permeability; Efflux transporter activity [1]. | Measure intracellular bioavailability (Fic); Use relevant cell types; Consider active transport mechanisms [1]. |
| High Biological Variation | Dynamic nature of metabolism; Sample type and sampling time differences; Uncontrolled pre-analytical factors [79]. | Standardize sampling procedures; Control for biological rhythms; Use appropriate statistical models to account for biological imprecision [79]. |
| Low Signal in Intracellular Staining | Inadequate fixation/permeabilization; Weakly expressed target paired with a dim fluorochrome [80]. | Optimize fixation/permeabilization protocol (e.g., ice-cold methanol); Use brightest fluorochrome (e.g., PE) for low-density targets [80]. |
| High Background in Flow Cytometry | Non-specific antibody binding; Presence of dead cells; Endogenous biotin interference [80]. | Block with BSA or serum; Use a viability dye; Gate out dead cells; Avoid biotinylated antibodies for intracellular staining [80]. |
Problem: Poor reproducibility of analytical data across different laboratories.
| Problem Area | Possible Causes | Recommendations |
|---|---|---|
| Method Reproducibility | Differences in instruments, reagents, personnel, or location between labs; Long time intervals between studies [79]. | Implement interlaboratory study (ILS) programs; Establish reproducibility conditions and precision statements [81]. |
| Variable Data Accuracy | Analytical methods with poor accuracy for specific elements/analytes; Lack of standardized reference materials [82]. | Use certified reference materials; Validate methods for problematic analytes (e.g., Pb, As, Sb); Participate in inter-laboratory comparisons [82]. |
Frequently Asked Questions (FAQs)
Q1: What is the difference between repeatability, intermediate precision, and reproducibility?
These terms describe measurement precision under different conditions [79].
- Repeatability: Variability measured under the same conditions, same instrument, and within a short time. This represents the smallest imprecision [79].
- Intermediate Precision: Variability measured within the same laboratory but over longer intervals (days/months) with changes in instruments, reagents, or personnel [79].
- Reproducibility: Variability measured between different laboratories. This represents the largest imprecision [79].
Q2: Why do some compounds show high biochemical potency but low cellular potency ("cell drop-off")?
This "cell drop-off" phenomenon often occurs because the compound cannot adequately access the intracellular target. The biochemical assay measures direct binding, while the cellular assay requires the compound to cross the cell membrane. A low intracellular bioavailability (Fic) means a smaller fraction of the dosed compound is available inside the cell to engage the target, leading to reduced observed potency [1].
Q3: How can I directly measure a compound's intracellular concentration to explain cell drop-off?
Traditional methods like logP or artificial membrane permeability are indirect estimates. A more direct approach uses a label-free method like RapidFire mass spectrometry to quantify the intracellular concentration of unbound drug molecules in whole cells. This provides a direct metric for intracellular bioavailability (Fic) and offers insights into cell permeability and accumulation [9] [1].
Q4: What are the best practices for fixing and permeabilizing cells for intracellular target staining?
- Fixation: Use methanol-free formaldehyde (e.g., 4%) and add immediately after treatment to cross-link proteins and preserve epitopes [80].
- Permeabilization: Use appropriate agents like Saponin, Triton X-100, or ice-cold 90% methanol. When using methanol, chill cells on ice first and add the methanol drop-wise while vortexing to prevent hypotonic shock and ensure homogeneous permeabilization [80].
Q5: How can our laboratory network formally assess and improve our inter-laboratory reproducibility?
Participate in or establish an Interlaboratory Study (ILS) Program, such as those coordinated by ASTM. These programs help laboratories [81]:
- Develop precision and bias statements for their test methods.
- Identify variability between laboratories (reproducibility) and within a single laboratory (repeatability).
- Produce a statistical summary and a formal research report that documents the study, which can be used to standardize methods across labs.
Data Presentation
Table 1: Inter-Laboratory Reproducibility of Bronze Composition Analysis
This table illustrates a real-world assessment of inter-laboratory reproducibility, showing how data accuracy can vary significantly for different elements [82].
| Analyte | Reproducibility Assessment |
|---|---|
| Cu, Sn, Fe, Ni | Results were fine (acceptable reproducibility) [82]. |
| Pb, Sb, Bi, Ag, Zn, Co, As, Mn, Al, Cd | Results were relatively poor (lower reproducibility and accuracy) [82]. |
Table 2: Key Concepts in Measurement Variation
Understanding this terminology is fundamental to diagnosing variability [79].
| Term | Definition | Clinical / Practical Implication |
|---|---|---|
| Accuracy | Closeness of agreement between a single measured value and a true reference value [79]. | Combines both bias and imprecision; reflected as total error [79]. |
| Trueness | Closeness of agreement between the average of repeated measurements and a reference value [79]. | Requires replicate measurements; reflects systematic error (bias) [79]. |
| Precision | Closeness of agreement between independent results under specified conditions [79]. | A measure of random error; inversely related to imprecision [79]. |
| Imprecision | The degree of inconsistency in repeated measurements of the same sample [79]. | Quantified by the Standard Deviation (SD); a large SD indicates low precision [79]. |
Experimental Protocols
Protocol 1: Assessing Intracellular Bioavailability (Fic)
Principle: This label-free methodology quantifies the fraction of extracellularly added drug that is available inside the cell in its unbound form, which is the fraction capable of engaging the intracellular target [1].
Methodology:
- Cell Preparation: Use cell types relevant to your target pharmacology (e.g., PBMCs for immunology, HeLa cells for general profiling) [1] [9].
- Compound Incubation: Incubate cells with the test compound at physiological conditions (e.g., 37°C) for a defined period.
- Separation and Washing: Rapidly separate cells from the incubation medium. Wash cells with a cold buffer to remove any extracellular compound adhering to the cell surface.
- Lysis and Analysis: Lyse the cells and analyze the lysate using a sensitive, high-throughput analytical technique such as RapidFire tandem mass spectrometry to determine the intracellular concentration of the unbound compound [9] [1].
- Calculation: Fic is determined by measuring the intracellular unbound fraction (fu,cell) and the cellular compound accumulation (Kp) [1].
Protocol 2: Conducting an Interlaboratory Study (ILS) for Standardization
Principle: An ILS involves multiple laboratories testing the same homogeneous samples using the same standardized method to quantify the method's repeatability (within-lab variation) and reproducibility (between-lab variation) [81].
Methodology (based on ASTM guidelines) [81]:
- Work Item Registration: Register the study with a coordinating body (e.g., ASTM).
- Sample Selection & Distribution: Secure identical, stable test samples and ship them to all participating laboratories alongside detailed instructions and data report forms.
- Data Collection: Each laboratory performs the test method on the provided samples and reports the raw data to the coordinating body.
- Statistical Analysis: The coordinating body analyzes the collated data using standardized statistical software (e.g., ASTM Statistical Software) to compute repeatability (r) and reproducibility (R).
- Report Generation: A draft research report is generated, including the precision and bias statement, a list of participating labs, raw data, and the statistical summary. This statement is then balloted for inclusion in the standard test method.
Diagnostic Diagrams
Measurement Conditions
Intracellular Bioavailability
The Scientist's Toolkit: Research Reagent Solutions
| Reagent / Material | Function in Experimental Context |
|---|---|
| Certified Reference Materials (CRMs) | Provides an accepted reference value to evaluate the accuracy (trueness and precision) of an analytical method, crucial for inter-laboratory comparisons [82]. |
| Ice-Cold Methanol (90%) | A common permeabilization agent for intracellular staining in flow cytometry; effectively exposes intracellular epitopes while preserving cell structure when used correctly (drop-wise addition to ice-chilled cells) [80]. |
| Formaldehyde (4%, Methanol-Free) | A cross-linking fixative used to preserve protein epitopes and cellular structure immediately after treatment. Methanol-free is recommended to prevent premature permeabilization and loss of intracellular proteins [80]. |
| Propidium Iodide (PI) / RNase Solution | A staining solution used in flow cytometry to label cellular DNA content, allowing for cell cycle analysis (distinguishing G0/G1, S, and G2/M phases) [80]. |
| Cyclosporine A | A pan-inhibitor of active transport processes (e.g., efflux pumps like P-glycoprotein). Used experimentally to investigate if active efflux is a mechanism limiting a compound's intracellular bioavailability (Fic) [1]. |
| Bovine Serum Albumin (BSA) / Normal Serum | Used as a blocking agent to reduce non-specific background staining in flow cytometry by occupying Fc receptors on immune cells, preventing non-specific antibody binding [80]. |
| Viability Dye (e.g., 7-AAD, Fixable Viability Dyes) | Distinguishes live cells from dead cells during flow cytometry analysis. Gating out dead cells is critical as they often exhibit high levels of non-specific antibody binding, which increases background noise [80]. |
A significant hurdle in modern drug discovery is the "cell drop-off" phenomenon, where a compound shows high affinity for its purified target in a biochemical assay but fails to elicit a cellular response [1]. This often occurs because the molecule cannot sufficiently cross the cell membrane to reach its intracellular target. In fact, analyses of drug discovery pipelines have revealed that uncertainty about target exposure is a major contributor to clinical trial failure [1]. This technical support guide addresses this core problem by outlining integrated experimental strategies to directly measure and overcome cell permeability barriers, thereby confidently validating engagement with intracellular targets.
Choosing the right assay depends on your specific experimental goal, whether it's initial target identification, validation of binding under physiological conditions, or quantifying intracellular compound concentration. The table below summarizes the key techniques discussed in this guide.
Table 1: Comparison of Key Target Engagement and Exposure Assays
| Method | Principle | Sample Type | Key Advantage | Primary Limitation |
|---|---|---|---|---|
| CETSA | Measures ligand-induced thermal stabilization of proteins [83] | Live cells or lysates [84] | Assesses target engagement in a physiological, intact cellular environment [83] [84] | Limited to proteins whose thermal stability is shifted by ligand binding [84] |
| DARTS | Detects ligand-induced protection from proteolysis [83] [84] | Cell lysates or purified proteins [84] | No chemical modification of the compound required; useful for proteins with minimal thermal shift [83] [84] | Lacks intact cellular context; requires careful protease optimization [84] |
| Fic Measurement | Quantifies the unbound intracellular drug concentration [1] | Live cells | Directly measures the fraction of bioavailable compound inside the cell, explaining potency gaps [1] | A separate methodology not directly proving target binding |
| Affinity-Based Profiling | Uses immobilized compound to pull down interacting proteins [83] | Lysates | High specificity for direct binders [83] | Requires chemical modification of the compound, which can alter its properties [83] |
Troubleshooting Guides & FAQs
FAQ 1: Why is my compound potent in a biochemical assay but inactive in a cellular assay?
This is a classic symptom of inadequate intracellular bioavailability (Fic) [1].
- Potential Cause: The compound has poor membrane permeability or is subject to active efflux transport, preventing it from reaching a sufficient concentration at the intracellular target site [1].
- Solution:
- Measure Intracellular Bioavailability: Determine the Fic value for your compound. This involves measuring the cellular compound accumulation (Kp) and the intracellular unbound fraction (fu,cell) to calculate the fraction of the external dose that is bioavailable inside the cell [1].
- Investigate Transport Mechanisms: Compare the Fic in the presence and absence of a pan-inhibitor of transport processes like cyclosporine A. A significant increase in Fic suggests the compound is an efflux transporter substrate [1].
- Use Cellular Target Engagement Assays: Employ CETSA in live cells to confirm whether the compound is engaging the target under physiological conditions. A negative CETSA result, despite good biochemical potency, strongly points to a permeability/efflux issue [83] [84].
FAQ 2: How do I choose between DARTS and CETSA for my target validation?
The choice hinges on the need for a physiologically relevant context versus flexibility and sensitivity to subtle conformational changes.
- Use CETSA when:
- You need to confirm the compound reaches and engages its target in a live cell, preserving the native cellular environment, including membrane potentials and protein complexes [83] [84].
- You require quantitative data on binding affinity (e.g., EC50) via ITDR-CETSA [83] [84].
- Your goal is unbiased, proteome-wide target identification (MS-CETSA) or profiling of off-target effects [83] [85].
- Use DARTS when:
- You are working with difficult-to-lyse cells or purified proteins.
- The protein-ligand interaction does not produce a significant thermal shift, as DARTS detects different, often more subtle, conformational changes [84].
- You are in early-stage discovery and need a quick, label-free method to confirm direct binding without specialized equipment [84].
- You are studying PROTAC molecules and want to confirm initial target engagement before degradation occurs [84].
FAQ 3: My CETSA experiment shows no thermal shift. What could be wrong?
A negative result does not always mean the compound does not bind.
- Troubleshooting Steps:
- Verify Binding in a Simpler System: Perform a DARTS experiment in a cell lysate. If DARTS is positive, the compound binds but may not induce thermal stabilization, or it may not be cell-permeable [84].
- Check the Cell Model: Ensure your cell model expresses the target protein at detectable levels.
- Optimize Experimental Conditions:
- Concentration: Use a sufficiently high compound concentration (e.g., 10-100x the biochemical IC50). For resistant cell lines, concentrations 500x the IC50 of sensitive lines may be needed to observe engagement [85].
- Incubation Time: Allow adequate time for the compound to enter cells and bind the target. Perform a time-course experiment (e.g., 1, 3, 5, 8 hours) to capture dynamic engagement [85].
- Confirm Assay Readout: Ensure your detection method (e.g., antibody for Western blot) is specific and sensitive enough for the target protein.
Table 2: Troubleshooting Common Experimental Issues
| Problem | Possible Causes | Suggested Solutions |
|---|---|---|
| No signal in CETSA/DARTS | Low target protein expression, insufficient compound concentration or incubation time, insensitive detection method. | Validate protein expression, perform dose-response and time-course experiments, use MS-based detection for broader coverage [83] [85]. |
| High background in DARTS | Protease concentration is too high, leading to non-specific degradation [84]. | Titrate the protease concentration to find the optimal level for limited proteolysis [84]. |
| Poor correlation between biochemical and cellular potency | Low intracellular bioavailability (Fic) due to poor permeability or active efflux; high cellular ATP levels competing with inhibitors [1]. | Measure Fic; use CETSA to confirm intracellular engagement; perform assays under physiologically relevant ATP levels [1]. |
| Inconsistent melting curves in CETSA | Poor cell lysis, temperature gradient inaccuracy, protein aggregation. | Standardize freeze-thaw lysis protocol; calibrate thermal cycler; include positive control compounds [83]. |
Detailed Experimental Protocols
Protocol 1: Western Blot-Based Cellular Thermal Shift Assay (WB-CETSA)
This protocol is used to validate target engagement for a specific protein in intact cells.
- Workflow Overview:
- Step-by-Step Methodology:
- Cell Treatment: Incubate live cells (e.g., in suspension or adhered) with your test compound or vehicle control for a predetermined time (e.g., 1-2 hours) at physiologically relevant conditions [83].
- Heating: Aliquot the cell suspensions into PCR tubes. Heat the samples across a gradient of temperatures (e.g., from 40°C to 65°C in 3-5°C increments) for 3-5 minutes using a precision thermal cycler [83].
- Cell Lysis: Lyse the cells immediately after heating using multiple rapid freeze-thaw cycles (e.g., in liquid nitrogen) [83].
- Fraction Separation: Centrifuge the lysates at high speed (e.g., 20,000 x g) to separate the soluble (folded) protein from the denatured and aggregated protein pellet [83].
- Detection and Analysis: Analyze the soluble protein fraction by SDS-PAGE and Western blotting using an antibody against your target protein. Quantify the band intensities and plot the fraction of soluble protein remaining versus temperature to generate a melting curve. A rightward shift (increase in Tm) in the compound-treated sample indicates thermal stabilization and target engagement [83].
Protocol 2: Isothermal Dose-Response CETSA (ITDR-CETSA)
This protocol is used to quantify the affinity of the drug-target interaction.
- Workflow Overview:
- Step-by-Step Methodology:
- Dose-Response Treatment: Incubate live cells with a series of concentrations of the test compound.
- Single Temperature Challenge: Heat all samples at a single temperature, typically chosen to be near the melting point (Tm) of the target protein from a previous full melt curve experiment [83].
- Processing and Detection: Lyse the cells, separate the soluble fraction by centrifugation, and detect the amount of remaining soluble target protein (via Western blot or MS).
- Data Analysis: Plot the amount of soluble protein (or the fraction of protein stabilized) against the logarithm of the compound concentration. Fit the data to a sigmoidal curve to determine the half-maximal effective concentration (EC50), which serves as a measure of cellular target engagement potency [83].
Protocol 3: Measuring Intracellular Bioavailability (Fic)
This protocol quantifies the unbound drug concentration inside cells.
- Step-by-Step Methodology:
- Cell Incubation: Incubate cells with the test compound at a known concentration for a specified time.
- Separation and Measurement: Separate the cells from the medium by centrifugation through a layer of oil or by rapid filtration. Measure the total drug concentration in the cells and the medium using a sensitive method like LC-MS/MS.
- Determine Unbound Fraction: Lyse a separate batch of cells and use equilibrium dialysis or ultrafiltration to determine the unbound fraction of the drug (fu,cell) in the cellular interior.
- Calculation: Calculate the intracellular bioavailability using the formula: Fic = (Intracellular unbound concentration) / (Extracellular concentration). A low Fic (<0.1) indicates poor intracellular exposure and explains a large cell drop-off [1].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions
| Reagent / Material | Function | Application Notes |
|---|---|---|
| Precision Thermal Cycler | Provides accurate and controlled heating of samples for CETSA. | Essential for generating reproducible thermal melt curves [83]. |
| Pronase / Thermolysin | Proteases used for limited proteolysis in DARTS. | Must be carefully titrated to avoid over-digestion; pronase is a mixture, thermolysin is specific [84]. |
| Mass Spectrometry System | For unbiased, proteome-wide detection of proteins in MS-CETSA and MS-DARTS. | Enables discovery of novel targets and off-target effects [83] [85]. |
| Specific Antibodies | Detect target proteins in Western blot-based CETSA and DARTS. | Critical for hypothesis-driven studies; requires validation for specificity [83]. |
| Cyclosporine A | Pan-inhibitor of efflux transporters like P-glycoprotein. | Used to investigate the role of active efflux in limiting intracellular compound accumulation [1]. |
| Cell Disruption Reagents | For lysis in DARTS and post-heating lysis in CETSA. | Freeze-thaw cycles are common for CETSA; mild detergents may be used for DARTS [83]. |
FAQs on Cell Permeability and Intracellular Assays
Q1: What are the primary causes of weak or no signal in intracellular target staining?
Weak or absent signal in flow cytometry can stem from several issues related to cell permeability and staining. Key causes and solutions include [86] [51]:
- Inadequate Permeabilization: The cell membrane may not be sufficiently permeabilized to allow antibody entry. Ensure you are using an appropriate permeabilization agent (e.g., saponin, Triton X-100, or ice-cold methanol) and that the protocol is optimized for your specific cell type and target [86].
- Low Target Expression: The intracellular protein may not be expressed at detectable levels. Always include a positive control of known antigen expression to confirm your assay is working [51].
- Large Fluorochrome Conjugates: The antibody-fluorochrome conjugate may be too large to efficiently enter the cell. Consider using smaller, brighter fluorochromes for low-abundance targets [86] [51].
- Antibody Incompatibility: The secondary antibody may not be compatible with the host species of the primary antibody. Verify species reactivity [51].
Q2: How can I reduce high background staining in my intracellular assays?
High background, which reduces the signal-to-noise ratio, is a common problem. To address it [86] [51] [87]:
- Optimize Antibody Concentration: Too much antibody can cause non-specific binding. Titrate your antibodies to find the optimal concentration [51].
- Block Effectively: Use Bovine Serum Albumin (BSA), Fc receptor blocking reagents, or normal serum from the secondary antibody host species to block non-specific binding sites [86] [87].
- Remove Dead Cells: Dead cells autofluoresce and bind antibodies non-specifically. Use a viability dye to gate out dead cells during analysis [86] [51].
- Wash Thoroughly: Include sufficient washes with detergents (e.g., Tween-20, Triton X-100) in the wash buffers to remove unbound antibodies [51].
Q3: What are the key trade-offs when choosing between advanced 3D models and traditional 2D cultures for permeability studies?
The choice of model system involves a direct trade-off between physiological relevance and throughput/scalability [17] [88].
- Physiological Relevance: Advanced 3D models (e.g., spheroids, organ-on-a-chip) better mimic the complex human tissue architecture, cell-cell interactions, and barrier functions found in vivo. For instance, Caco-2/HT29-MTX co-cultures replicate the intestinal mucosa more accurately than Caco-2 monocultures [17].
- Throughput & Cost: Traditional 2D cultures and simpler 3D models (like liver spheroids) are more scalable, cost-effective, and have faster turnaround times. In contrast, complex Organ-on-Chip systems can be up to 100 times more expensive and require longer preparation times, making them less suitable for high-throughput screening [88].
Troubleshooting Guides
Guide 1: Troubleshooting Weak Signal in Flow Cytometry
| Problem | Possible Cause | Recommended Solution |
|---|---|---|
| Weak/No Signal | Inadequate fixation/permeabilization | Use fresh, methanol-free formaldehyde for fixation. For permeabilization, add ice-cold methanol drop-wise while gently vortexing [86]. |
| Low target expression | Incorporate a positive control. Use the brightest fluorochrome (e.g., PE) for the lowest density target [86] [51]. | |
| Large fluorochrome conjugate | Use smaller fluorochromes for better cell entry, especially for nuclear targets [86]. | |
| Incompatible laser/PMT settings | Ensure the flow cytometer's laser wavelength and detector settings match the fluorochrome's excitation/emission spectra [86]. | |
| High Background | Non-specific antibody binding | Block with BSA, serum, or Fc receptor blockers. Include an isotype control. Perform additional wash steps [86] [51]. |
| Presence of dead cells | Use a viability dye (e.g., PI, 7-AAD) to gate out dead cells during analysis [86]. | |
| High antibody concentration | Titrate antibody to find the optimal concentration. Reduce amount used [51]. | |
| Suboptimal Scatter | Clogged flow cell | Unclog the instrument per manufacturer's instructions (e.g., run 10% bleach followed by dH₂O) [86]. |
| Incorrect instrument settings | Ensure proper instrument settings are loaded. Use fresh, healthy cells to set FSC and SSC [86] [51]. |
Guide 2: Selecting Models for Permeability and Toxicity Studies
| Model System | Key Advantages (Throughput/Scalability) | Key Limitations (Physiological Relevance) | Ideal Use Case |
|---|---|---|---|
| Caco-2 Monoculture | Standardized, reproducible, moderate throughput [17]. | Extended cultivation time (~21 days), lacks mucosal layer [17]. | Early-stage, high-throughput permeability screening of small molecules [17]. |
| Parallel Artificial Membrane Permeability Assay (PAMPA) | Very high throughput, low cost, simple operation [17]. | Non-cell-based, does not capture active transport or metabolism [17]. | Ultra-high-throughput initial lipophilicity and passive permeability ranking [17]. |
| 3D Liver Spheroids | Good balance of relevance and throughput; cost-effective; highly reproducible [88]. | Lack dynamic flow, may not capture all immune interactions [88]. | Routine DILI (Drug-Induced Liver Injury) assessment and mechanistic studies [88]. |
| Organ-on-a-Chip (OoC) | High physiological relevance; dynamic flow; mechanical forces; can model multi-tissue interactions [17] [88]. | Very high cost (up to 100x more); low throughput; complex operation; requires large cell numbers [88]. | Late-stage, mechanistic investigation of complex biological questions and human-specific pathways [88]. |
| Induced Pluripotent Stem Cells (iPSCs) | Patient-specific data; can model human-specific biology and genetic diseases; potential for high relevance [17] [89]. | Can be variable; differentiation and culture can be lengthy and complex [17] [89]. | Personalized medicine, disease modeling (e.g., neurodegenerative diseases) [89]. |
Experimental Protocols
Protocol 1: Standard Intracellular Staining for Flow Cytometry
This protocol is optimized for detecting intracellular proteins like transcription factors or cytokines [86].
Key Research Reagent Solutions:
- Fixation Buffer: 4% methanol-free formaldehyde in PBS. Cross-links and preserves cell structures [86].
- Permeabilization Buffer: 90% ice-cold methanol or buffers containing saponin/Triton X-100. Dissolves lipids to create holes in the membrane [86].
- Staining Buffer: PBS containing 0.5-1% BSA. Used to dilute antibodies and block non-specific binding [86].
- Fc Receptor Blocking Solution: Normal serum or commercial blockers. Reduces non-specific antibody binding via Fc receptors [86] [87].
- Viability Dye: e.g., Propidium Iodide (PI) or 7-AAD. Distinguishes live from dead cells [86].
Methodology:
- Harvest and Wash: Collect cells and wash once with cold flow cytometry staining buffer.
- Surface Staining (Optional): If staining surface markers, incubate with directly conjugated antibodies for 20-30 minutes on ice. Wash twice with cold buffer.
- Fixation: Resuspend cell pellet in 4% formaldehyde. Fix for 15-20 minutes at room temperature. Critical: Use methanol-free formaldehyde to prevent loss of intracellular proteins [86].
- Permeabilization: Centrifuge and thoroughly resuspend cell pellet in ice-cold 90% methanol. Critical: Add methanol drop-wise while gently vortexing to ensure homogeneous permeabilization and prevent hypotonic shock. Incubate for at least 30 minutes on ice. Cells can be stored at -20°C in methanol at this stage [86].
- Intracellular Staining: Wash cells twice with permeabilization buffer (e.g., 0.1% Saponin in staining buffer) to remove methanol. Resuspend cells in permeabilization buffer containing pre-titrated fluorescently-labeled antibodies. Incubate for 30-60 minutes at room temperature in the dark.
- Final Wash and Acquisition: Wash cells twice with permeabilization buffer, then once with staining buffer. Resuspend in an appropriate volume of staining buffer and analyze on the flow cytometer.
Protocol 2: Establishing a Caco-2 Model for Intestinal Permeability
This protocol outlines the setup of a traditional 2D Caco-2 model for predicting drug absorption [17].
Key Research Reagent Solutions:
- Caco-2 Cell Line: Human colorectal adenocarcinoma cells. Differentiate into enterocyte-like cells [17].
- Electrospun Nanofiber Scaffolds: Synthetic scaffolds used to enhance the performance and accelerate the formation of polarized cell monolayers [17].
- Accelerated Differentiation Media: Specialized media formulations that reduce the time required for Caco-2 cells to fully differentiate [17].
- HT29-MTX Cell Line: Mucin-producing goblet cells. Used in co-culture with Caco-2 to create a more physiologically relevant model with a mucosal layer [17].
Methodology:
- Cell Seeding: Culture Caco-2 cells in standard DMEM media. Seed onto a semi-permeable filter insert (e.g., Transwell) at a high density (~60,000 cells/cm²).
- Differentiation: Allow cells to grow for 14-21 days, changing the media every 2-3 days. During this time, the cells proliferate, become confluent, and spontaneously differentiate into a polarized monolayer with tight junctions and brush border enzymes.
- Validation: Monitor the formation of tight junctions by regularly measuring the Transepithelial Electrical Resistance (TEER). The model is ready for experiments when TEER values plateau at a high level (typically > 300 Ω·cm²).
- Permeability Assay: Add the test compound to the apical (donor) compartment. Sample from the basolateral (receiver) compartment at regular time points over a few hours.
- Analysis: Quantify the compound in the receiver compartment using analytical methods like HPLC or LC-MS. Calculate the apparent permeability coefficient (Papp).
Signaling Pathways and Workflows
Intracellular Staining Workflow
Model System Selection Logic
Conclusion
Overcoming cell permeability challenges requires an integrated, multidisciplinary strategy that combines sophisticated experimental models with computational prediction and thoughtful compound design. The evolution from traditional monolayer assays to complex 3D models and organ-on-a-chip systems provides increasingly physiologically relevant platforms, while AI-driven tools offer unprecedented capability for virtual screening. Success in intracellular target engagement demands careful validation across multiple assay formats and acknowledgment of the inherent trade-offs between throughput and biological complexity. Future progress will likely emerge from enhanced bio-relevant screening models, more accurate computational predictors that incorporate membrane interaction dynamics, and innovative delivery technologies that expand the chemical space of druggable intracellular targets. By systematically addressing permeability barriers, researchers can unlock previously inaccessible biological targets and accelerate the development of novel therapeutics for a wide range of diseases.
References
- 1. Prediction of intracellular exposure bridges the gap between ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5544291/]
- 2. Thermal shift assays in drug discovery [https://www.sciencedirect.com/science/article/abs/pii/S1056871925008007]
- 3. Improving the Accuracy of Permeability Data to Gain ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11278619/]
- 4. Article Development of an intracellular quantitative assay to ... [https://www.sciencedirect.com/science/article/pii/S2451945621003585]
- 5. Trends in Molecular Properties, Bioavailability, and ... [https://pubmed.ncbi.nlm.nih.gov/36752336/]
- 6. Cell permeability beyond the rule of 5 [https://www.sciencedirect.com/science/article/abs/pii/S0169409X16300990]
- 7. Effects of intramolecular hydrogen bonds on lipophilicity [https://www.sciencedirect.com/science/article/abs/pii/S0928098719300260]
- 8. Impact of Stereospecific Intramolecular Hydrogen Bonding ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3968888/]
- 9. Direct Measurement of Intracellular Compound ... [https://www.sciencedirect.com/science/article/pii/S2472555222070642]
- 10. Caco-2 Cell Tips for Reliable Intestinal Permeability Models [https://mimetas.com/resources/7-ways-caco-2-cells-can-make-or-break-your-intestinal-permeability-models]
- 11. Conquering the beyond Rule of Five Space with an Optimized ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11280027/]
- 12. Inside the Ring Series 7: Beyond Rule of Five for ... [https://www.linkedin.com/pulse/inside-ring-series-7-beyond-rule-five-macrocycles-just-wentao-guo-jnrfc]
- 13. Taking on bRo5 Compounds [https://www.tabletscapsules.com/3641-Technical-Articles/619821-Taking-on-bRo5-Compounds/]
- 14. Systematic benchmarking of 13 AI methods for predicting ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-025-01083-4]
- 15. Cyclic peptide membrane permeability prediction using ... [https://www.frontiersin.org/journals/bioinformatics/articles/10.3389/fbinf.2025.1566174/full]
- 16. Prodrug Approach as a Strategy to Enhance Drug Permeability [https://pmc.ncbi.nlm.nih.gov/articles/PMC11946379/]
- 17. Advancements in techniques for analyzing cell permeability [https://www.sciencedirect.com/science/article/pii/S2405844025022236]
- 18. Navigating Drug Design in 'Beyond the Rule of 5' Landscape [https://www.acdlabs.com/blog/navigating-drug-design-in-beyond-the-rule-of-5-landscape/]
- 19. Using in vitro ADME Data for Lead Compound Selection [https://pmc.ncbi.nlm.nih.gov/articles/PMC9843724/]
- 20. Comparison of MDCK-MDR1 and Caco-2 Cell-Based ... [https://www.creative-bioarray.com/support/comparison-of-mdck-mdr1-and-caco-2-cell-based-permeability-assays.htm]
- 21. Pitfalls in evaluating permeability experiments with Caco-2/ ... [https://www.sciencedirect.com/science/article/pii/S0928098724000101]
- 22. Predicting apparent passive permeability of Caco-2 and ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0190319]
- 23. Overcoming the cell membrane to target intracellular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10369770/]
- 24. Strategies to promote permeation and vectorization ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8341117/]
- 25. Meta-Analysis of Permeability Literature Data Shows ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11881145/]
- 26. 3D organ-on-a-chip: The convergence of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9720174/]
- 27. A guide to the organ-on-a-chip [https://www.nature.com/articles/s43586-022-00118-6]
- 28. Advances in organoid-on-a-chip for recapitulation of ... [https://www.sciencedirect.com/science/article/abs/pii/S1369702125000355]
- 29. Flow Cytometry Troubleshooting Guide [https://www.cellsignal.com/learn-and-support/troubleshooting/flow-cytometry-troubleshooting-guide?srsltid=AfmBOopGiofyHY6BY1KRofdLWYxcgYMmudWjUpSeV8FZiPI7xYxZ_hTF]
- 30. Prediction of Permeability and Efflux Using Multitask ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12631307/]
- 31. Advances in Computational Approaches for Estimating ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10673305/]
- 32. Orion Permeability Workflow [https://www.eyesopen.com/orion/permeability]
- 33. Determination of In Vitro Membrane Permeability by ... [https://iv.iiarjournals.org/content/33/6/1767]
- 34. Emerging Target Discovery Strategies Drive the Decoding of... [https://pubmed.ncbi.nlm.nih.gov/40873643/]
- 35. Frontiers | Celastrol : A Review of Useful Strategies Overcoming its... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2020.558741/full]
- 36. From Physicochemical Constraints to Clinical Prospects of Celastrol ... [https://www.dovepress.com/from-physicochemical-constraints-to-clinical-prospects-of-celastrol-ch-peer-reviewed-fulltext-article-IJN]
- 37. Assay Guidance Manual - NCBI Bookshelf - NIH [https://www.ncbi.nlm.nih.gov/books/NBK53196/]
- 38. Flow Cytometry Troubleshooting Guide [https://www.cellsignal.com/learn-and-support/troubleshooting/flow-cytometry-troubleshooting-guide?srsltid=AfmBOooXlpG0cxnDbgEfH39r_oQ2oWFPlNlHJeh5PK_d0OYPTtrJ_NUX]
- 39. Frontiers | Assessment of drug using a small airway... permeability [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1621775/full]
- 40. Commercially Available Cell -Free Permeability Tests for Industrial... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9964961/]
- 41. Troubleshooting Guide for Common CRISPR-Cas9 Editing ... [https://synapse.patsnap.com/article/troubleshooting-guide-for-common-crispr-cas9-editing-problems]
- 42. Miniaturized scalable arrayed CRISPR screening in ... [https://www.nature.com/articles/s41598-025-13532-z]
- 43. CRISPR Screen Analysis: Expert Troubleshooting Q&A [https://www.ubigene.us/application/crispr-screening-data-analytics-faq.html]
- 44. Validation of a Miniaturized Permeability Assay Compatible ... [https://pubmed.ncbi.nlm.nih.gov/31578372/]
- 45. Resveratrol and pterostilbene: A comparative overview ... [https://japsonline.com/abstract.php?article_id=2952&sts=2]
- 46. Biological actions and molecular effects of resveratrol ... [https://www.sciencedirect.com/science/article/pii/S1021949816300965]
- 47. Comparative study of lipophilicity, cell membrane ... [https://www.sciencedirect.com/science/article/abs/pii/S0955286325002578]
- 48. Pterostilbene and resveratrol: Exploring their protective ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12022656/]
- 49. Resveratrol and pterostilbene: A comparative overview of ... [https://www.semanticscholar.org/paper/Resveratrol-and-pterostilbene%3A-A-comparative-of-and-Chan-Wong/5e756d4acd5e460c6d0f3c3dba5b318ff232052d]
- 50. Flow Cytometry Troubleshooting Guide [https://www.cellsignal.com/learn-and-support/troubleshooting/flow-cytometry-troubleshooting-guide?srsltid=AfmBOopeHpMIAKY19U8te4lH-ArTGbmR5INScEqcFDkTfjHHxwJ8arCd]
- 51. Troubleshooting of Intracellular Staining Flow Cytometry [https://www.antibody-creativebiolabs.com/troubleshooting-of-intracellular-staining-flow-cytometry.htm]
- 52. Holland-Frei Cancer Medicine - NCBI Bookshelf [https://www.ncbi.nlm.nih.gov/books/NBK13555/]
- 53. A Systematic Review and Classification of the Effects of P ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12158833/]
- 54. Nanotherapeutics approaches to overcome P-glycoprotein- ... [https://www.sciencedirect.com/science/article/abs/pii/S1549963421001374]
- 55. P-Glycoprotein: One Mechanism, Many Tasks and the ... [https://www.frontiersin.org/journals/oncology/articles/10.3389/fonc.2020.576559/full]
- 56. Bypassing P-Glycoprotein Drug Efflux Mechanisms [https://pmc.ncbi.nlm.nih.gov/articles/PMC4600488/]
- 57. EFFLUX TRANSPORTERS: Newly Appreciated Roles in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3160781/]
- 58. Liposome-Based Drug Delivery Systems: From Laboratory ... [https://www.mdpi.com/2305-7084/9/3/56]
- 59. Strategic advances in liposomes technology: translational ... [https://jnanobiotechnology.biomedcentral.com/articles/10.1186/s12951-025-03660-z]
- 60. Advances in Liposomal Drug Delivery - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC12298344/]
- 61. LNP characterization guidelines: Size, PDI, Morphology [https://insidetx.com/resources/reviews/lnp-and-liposomes-characterization-guidelines/]
- 62. Challenges in Development of Nanoparticle-Based ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3326161/]
- 63. Nanoparticles Modified with Cell-Penetrating Peptides [https://www.mdpi.com/1422-0067/21/7/2536]
- 64. Microfluidic-based nanocarriers for overcoming biological ... [https://pubs.rsc.org/en/content/articlehtml/2025/nr/d5nr02322j?page=search]
- 65. Creating Consistent Results by Managing your Flow ... [https://blog.td2inc.com/creating-consistent-results-by-managing-your-flow-cytometry-data]
- 66. Assessment Using 5-day Cultured Caco-2 Permeability Monolayers Cell [https://experiments.springernature.com/articles/10.1007/978-1-62703-742-6_4?error=cookies_not_supported&code=26be2fbb-c274-4b13-b204-9e3dd4576492]
- 67. Flow Cytometry Troubleshooting Guide [https://www.cellsignal.com/learn-and-support/troubleshooting/flow-cytometry-troubleshooting-guide?srsltid=AfmBOoqZ3N8Csk72PbLM-RjpD6AWW7ja1ILpBfXb-g5PENXfP1iarHdA]
- 68. Caco-2 Cells Protocol – MedTech Barcelona Permeability Assay [https://medtechbcn.com/readycell/blog/caco-2-permeability-protocol/]
- 69. Peptide Optimizing : Insights from Our GRC... Permeability Assays [https://www.linkedin.com/pulse/optimizing-peptide-permeability-assays-insights-from-our-grc-e01ee]
- 70. of Optimization permeabilization in electron flow based... cell [https://pubmed.ncbi.nlm.nih.gov/34656699/]
- 71. An Update on Flow Cytometry Analysis of Hematological ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12190783/]
- 72. Advancing Drug Discovery: The Power of Flow Cytometry ... [https://kcasbio.com/blogs/advancing-drug-discovery-the-power-of-flow-cytometry-in-compound-screening/]
- 73. QSPR model for Caco-2 cell permeability prediction using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9058322/]
- 74. CaliciBoost: Performance-Driven Evaluation of Molecular ... [https://arxiv.org/html/2506.08059v1]
- 75. Employing Automated Machine Learning (AutoML) Methods to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12004515/]
- 76. Improving In Vitro–In Vivo Correlation (IVIVC) for Lipid ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12567226/]
- 77. Revisiting the in-vitro and in-vivo considerations for in- ... [https://www.sciencedirect.com/science/article/abs/pii/S0168365923004029]
- 78. Guidelines for Comparing in Silico, in Vitro, and in Vivo ... [https://pubmed.ncbi.nlm.nih.gov/39823383/]
- 79. Are Your Laboratory Data Reproducible? The Critical Role of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11996692/]
- 80. Flow Cytometry Troubleshooting Guide [https://www.cellsignal.com/learn-and-support/troubleshooting/flow-cytometry-troubleshooting-guide?srsltid=AfmBOoqfZk602Z9bzo498wM2MQka5GpALZWXVjuIomQ3K1afyGhBqRBO]
- 81. Interlaboratory Study Program [https://www.astm.org/membership-participation/technical-committees/interlaboratory-studies-program]
- 82. Evaluating accuracy and inter-laboratory reproducibility of ... [https://www.sciencedirect.com/science/article/pii/S266713602300016X]
- 83. Applications of the Cellular Thermal Shift Assay to Drug ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12072176/]
- 84. DARTS vs CETSA: Choosing the Right Assay for Target ... [https://www.creative-proteomics.com/resource/darts-vs-cetsa-choosing-the-right-assay-for-target-validation.htm]
- 85. MS CETSA deep functional proteomics uncovers DNA ... [https://www.nature.com/articles/s41467-025-59505-8]
- 86. Flow Cytometry Troubleshooting Guide [https://www.cellsignal.com/learn-and-support/troubleshooting/flow-cytometry-troubleshooting-guide?srsltid=AfmBOorzNVZpSAiKOefjYdIlXPd04-vpDru6moxNQMfWhQgRtHPPanFm]
- 87. IHC Troubleshooting Guide [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/ihc-troubleshooting-guide.html]
- 88. DILI Assessment | Navigating the Trade-offs in 3D In Vitro [https://insphero.com/navigating-dili-assessment-trade-offs/]
- 89. Advanced 3D biomaterials and bioprinting strategies for in ... [https://www.sciencedirect.com/science/article/pii/S2949723X25000212]