Molecular Cloning and Recombinant DNA Technology: Revolutionizing Neuroscience Research and Therapeutics
This article provides a comprehensive overview of how molecular cloning and recombinant DNA technologies are fundamentally advancing neuroscience.
Molecular Cloning and Recombinant DNA Technology: Revolutionizing Neuroscience Research and Therapeutics
Abstract
This article provides a comprehensive overview of how molecular cloning and recombinant DNA technologies are fundamentally advancing neuroscience. Tailored for researchers and drug development professionals, it explores the foundational principles of manipulating neural DNA, details cutting-edge methodologies from PCR cloning to gene synthesis, and offers practical guidance for troubleshooting and optimizing experiments. It further covers critical validation techniques and comparative analyses of cloning methods, highlighting their direct application in studying brain function, modeling disease, and developing novel therapeutic strategies for neurological disorders.
The Building Blocks of the Brain: Core Principles of Recombinant DNA Technology in Neuroscience
Recombinant DNA technology, fundamentally, is a set of molecular techniques that allow for the assembly of DNA molecules from different sources into a single, novel recombinant DNA molecule, which can then be replicated and propagated in a host organism [1] [2]. This capability, born from the understanding of bacterial defense mechanisms, has revolutionized all fields of biology, including neuroscience.
The technology's foundation lies in the restriction-modification system, a bacterial immune system that protects against invading viruses (bacteriophages) [2]. In this system, a restriction enzyme cleaves foreign DNA at specific sequences, while a methylase modifies the host's own DNA at the same sequences, protecting it from cleavage [3]. The discovery and isolation of these sequence-specific restriction enzymes provided the precise "molecular scissors" necessary for the birth of genetic engineering [2] [3]. This article provides detailed application notes and protocols for leveraging this powerful technology in modern neuroscience research.
Core Principles and Key Reagents
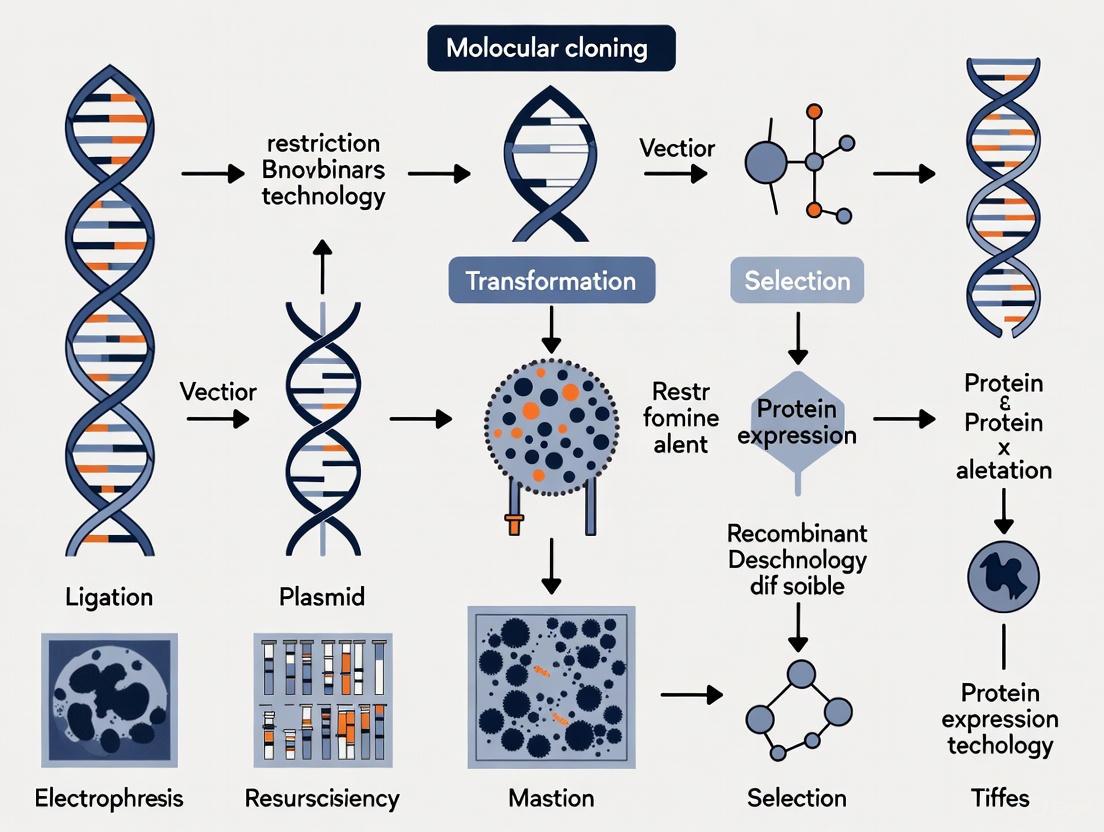
The Molecular Cloning Workflow
The standard workflow for creating a recombinant DNA molecule involves several key steps, each requiring specific reagents and techniques [1] [2].
The Scientist's Toolkit: Essential Research Reagents
Successful recombinant DNA experiments rely on a core set of reagents and biological tools.
Table 1: Key Research Reagent Solutions for Molecular Cloning
| Reagent/Biological Tool | Function/Description | Key Applications in Neuroscience |
|---|---|---|
| Restriction Enzymes (e.g., EcoRI, HindIII) | Enzymes that cut DNA at specific palindromic sequences, generating defined fragments [1] [2]. | Excision of a gene of interest (GOI) from genomic DNA for cloning into an expression vector. |
| DNA Ligase (e.g., T4 DNA Ligase) | Enzyme that catalyzes the formation of a phosphodiester bond between the 3'-hydroxyl and 5'-phosphate ends of DNA, joining fragments [1] [2]. | Ligation of a neuronal promoter sequence into a plasmid vector upstream of a reporter gene. |
| Cloning Vectors (e.g., Plasmids) | Small, circular DNA molecules that autonomously replicate in a host cell. Contain an Origin of Replication (Ori), selectable marker, and multi-cloning site [1] [3]. | Propagation and amplification of DNA encoding a neuroreceptor subunit. |
| Expression Vectors | Specialized vectors containing strong promoters (e.g., CMV, CAG) and other regulatory elements to drive high-level protein production in host cells [3]. | Overexpression of a channelrhodopsin protein in neuronal cultures for optogenetics experiments. |
| Competent Cells | Host cells (typically E. coli) treated to become permeable to foreign DNA, enabling transformation via heat shock or electroporation [1] [2]. | Amplification of plasmid DNA for in vivo transfection or viral packaging. |
| Polymerase Chain Reaction (PCR) | A laboratory method for amplifying a specific DNA sequence exponentially using primers and a DNA polymerase [1]. | Amplification of a cDNA template for a synaptic protein from a brain-derived RNA sample. |
Modern DNA Assembly Strategies
While traditional restriction enzyme cloning is foundational, several advanced methods have been developed to overcome its limitations, such as dependence on restriction sites and the potential for unwanted "scar" sequences [1] [3].
Table 2: Comparison of Modern DNA Assembly Strategies
| Method | Principle | Key Advantage | Limitation | Typical Efficiency |
|---|---|---|---|---|
| Golden Gate Assembly | Uses Type IIS restriction enzymes, which cut outside their recognition site, allowing for seamless, scarless assembly of multiple fragments in a single reaction [1] [3]. | High efficiency and fidelity for multi-fragment assembly; seamless. | Requires careful design of fragment overhangs. | High (>90% positive clones common) |
| Gibson Assembly | An isothermal, single-reaction method that uses a 5' exonuclease, a DNA polymerase, and a DNA ligase to assemble multiple overlapping DNA fragments [1]. | Seamless and method-agnostic; excellent for large DNA constructs. | Requires PCR to generate homologous overlaps, risking mutation. | High |
| Gateway Cloning | A site-specific recombination-based system that uses bacteriophage λ attachment (att) sites and LR/BP Clonase enzymes to shuttle DNA sequences between vectors [1] [3]. | Highly efficient and standardized; allows easy transfer of GOI between different vector systems. | Proprietary system; leaves a short recombination "scar" sequence. | Very High |
| TA Cloning | Leverages the terminal transferase activity of some DNA polymerases (e.g., Taq) which adds a single deoxyadenosine (A) to the 3' end of PCR products. These are ligated into a vector with a complementary T-overhang [1]. | Simple and rapid for cloning PCR products. | Non-directional; not suitable for multi-fragment assembly. | Moderate |
The following diagram illustrates the core mechanisms of two widely used seamless cloning methods.
Application Notes & Protocols for Neuroscience
Protocol: Cloning a Neuronal Gene cDNA into an Expression Vector using Gibson Assembly
This protocol is ideal for creating a plasmid to express a protein in neuronal cell lines or in vivo.
Materials:
- Gene of Interest (GOI): cDNA for a neuronal receptor (e.g., GluA1 AMPA receptor subunit).
- Linearized Vector: Mammalian expression vector with a neuronal promoter (e.g., synapsin).
- Gibson Assembly Master Mix: Commercial kit (e.g., from New England Biolabs).
- Competent E. coli: High-efficiency strains (e.g., NEB 5-alpha).
- Antibiotics: Appropriate for vector selection.
Method:
- Amplify Insert with Homology Arms:
- Design PCR primers to amplify the GluA1 cDNA. The forward and reverse primers must include 20-40 bp extensions at their 5' ends that are homologous to the sequence flanking the insertion site in the linearized vector.
- Perform a high-fidelity PCR to generate the insert. Purify the PCR product using silica column or magnetic bead-based purification.
Prepare Linearized Vector:
- If using a restriction enzyme, digest the vector plasmid and gel-purify the linear backbone.
- Alternatively, use a PCR-based method to linearize the vector. The final vector must lack the GOI but contain the homologous regions for assembly.
Gibson Assembly Reaction:
- Set up the following reaction on ice:
- 2x Gibson Assembly Master Mix: 10 µL
- Linearized Vector: 50-100 ng (optimize molar ratio)
- Insert DNA: Molar ratio of 2:1 to 3:1 (insert:vector)
- Nuclease-free water to 20 µL
- Mix gently and incubate at 50°C for 15-60 minutes.
- Set up the following reaction on ice:
Transformation:
- Transform 2-5 µL of the assembly reaction into 50 µL of chemically competent E. coli via heat shock, or 1 µL into electrocompetent cells via electroporation [2].
- Add recovery medium and incubate with shaking for 1 hour.
- Plate onto LB agar plates containing the appropriate antibiotic.
Screening and Validation:
- After overnight growth, pick colonies for screening via colony PCR.
- Inoculate positive clones into liquid culture for plasmid DNA isolation (miniprep).
- Verify the clone by diagnostic restriction digest and Sanger sequencing of the entire insert and junctions.
Application Note: Recombinant DNA in the BRAIN Initiative
The BRAIN Initiative aims to revolutionize our understanding of the brain, and recombinant DNA technology is the engine driving this progress [4]. Its applications are central to achieving the initiative's primary goals:
Cell Type Census: Recombinant tools are used to create transgenic reporter lines where cell-type-specific promoters (e.g., for parvalbumin interneurons) drive the expression of fluorescent proteins (e.g., GFP). This allows for the identification, isolation, and morphological characterization of specific neuronal populations in the complex brain environment [4].
Mapping Neural Circuits: The development of recombinant viral vectors (e.g., Adeno-associated virus - AAV) encoding tract-tracers like the Wheat Germ Agglutinin (WGA) or engineered variants of the rabies virus enables the mapping of synaptic connections between different brain regions with high precision [4].
Monitoring and Manipulating Neural Activity: Recombinant DNA technology underpins optogenetics and chemogenetics. Genes for light-sensitive ion channels (e.g., Channelrhodopsin-2) or engineered GPCR receptors (e.g., DREADDs) are cloned into viral vectors and delivered to specific brain regions. This allows researchers to precisely activate or inhibit defined neuronal populations in behaving animals, establishing causal links between neural activity and behavior [4].
Quantitative Analysis in Gene Expression
Following the cloning and application of recombinant constructs, quantifying gene expression changes is critical. Quantitative PCR (qPCR) is the standard method for this.
Table 3: Comparison of qPCR Data Analysis Methods [5]
| Analysis Method | Description | Relative Accuracy (RE) | Relative Precision (CV) |
|---|---|---|---|
| Simple Linear Regression (SLR) | Standard linear regression of log fluorescence vs. cycle number in the exponential phase. | 0.233 (Taking-Difference) | 26.80% (Taking-Difference) |
| Weighted Linear Regression (WLR) | Linear regression that accounts for heteroscedasticity by applying a weight factor (e.g., reciprocal of variance). | 0.123 (Taking-Difference) | 19.50% (Taking-Difference) |
| Linear Mixed Model (LMM) | Accounts for both fixed and random effects, suitable for data with repeated measures or hierarchical structure. | 0.216 (Taking-Difference) | 20.40% (Taking-Difference) |
| Weighted Linear Mixed Model (WLMM) | Combines the advantages of weighting and mixed models for complex experimental designs. | Most precise | Most precise |
Key Finding: A study comparing analysis models found that preprocessing qPCR data using the "taking-the-difference" approach (subtracting fluorescence of cycle n-1 from cycle n) reduced background estimation error and improved accuracy compared to the standard background subtraction method [5]. Furthermore, weighted models (WLR, WLMM) consistently provided more accurate and precise estimations of the initial DNA amount, with the weighted linear mixed model being the most robust for complex data sets [5].
Recombinant DNA technology, born from the study of bacterial defense, has become an indispensable tool in neuroscience. From the foundational techniques of restriction and ligation to modern, seamless assembly methods, this technology enables the precise dissection of neural circuits, the identification of cell types, and the causal manipulation of brain activity. As outlined in these application notes and protocols, the continued refinement of these methods, coupled with rigorous quantitative analysis, drives progress toward the ambitious goals of the BRAIN Initiative and the broader quest to understand the brain in health and disease.
Molecular cloning is a cornerstone of modern neuroscience, enabling the study of neural gene function, the development of disease models, and the creation of advanced tools for neuronal manipulation. This application note provides a detailed framework for employing recombinant DNA technology in neural research, focusing on the critical triad of restriction enzymes, vectors, and host organisms.
The Core Toolkit for Molecular Cloning in Neuroscience
The fundamental process of molecular cloning involves inserting a gene of interest into a plasmid vector, which is then replicated within a host organism to produce multiple copies. This recombinant DNA technology has been revolutionary for isolating and studying individual genes [6] [7].
Restriction Enzymes: The Molecular Scissors
Restriction enzymes are proteins that recognize and cleave DNA at specific sequences, functioning as a bacterial defense system against foreign DNA [6] [8]. They are indispensable for cutting DNA fragments for cloning.
Types and Activities: The most common types are Type IIP enzymes, which recognize palindromic sequences and cut within that sequence. Their cleavage produces three types of ends [7]:
- 5' protruding ends: A 5' stretch of unpaired DNA.
- 3' protruding ends: A 3' overhang of unpaired DNA.
- Blunt ends: No overhangs.
Type IIS restriction enzymes, such as BsaI and BsmBI, cleave DNA at a defined distance from their recognition site and are the basis for powerful, seamless assembly methods like Golden Gate cloning [3] [1].
Table 1: Common Restriction Enzymes and Their Properties in Cloning
| Enzyme | Type | Recognition Sequence | End Type | Common Application in Neuroscience |
|---|---|---|---|---|
| EcoRI [8] | IIP | GAATTC | 5' overhang | Traditional gene cloning into plasmid vectors. |
| BamHI [8] | IIP | GGATCC | 5' overhang | Insertion of genes into expression vectors. |
| SmaI [8] | IIP | CCCGGG | Blunt | Cloning where orientation is not critical. |
| BsaI [1] | IIS | GGTCTC(N)₁ | 5' overhang | Golden Gate assembly for seamless, multi-part construct building. |
Vectors: The Molecular Delivery Vehicles
A vector is a DNA molecule that carries foreign genetic material into a host cell. Essential features include an origin of replication (Ori) for in vivo amplification, a selectable marker (e.g., antibiotic resistance) for selective growth, and a multiple cloning site (MCS) for insertion of the DNA fragment [7] [1].
Specialized Vectors for Neuroscience: Beyond standard cloning plasmids, neuroscience research often requires sophisticated vectors for gene delivery and expression in neural cells and tissues.
- Adeno-Associated Virus (AAV) Vectors: AAV has become a pivotal tool for gene therapy and neural circuit manipulation due to its safety profile and ability to transduce non-dividing cells, including neurons [9]. A key advancement is the use of cell-type-specific enhancers to control transgene expression. A recent study demonstrated that transcriptional crosstalk between co-transduced AAV genomes enables cell-type-specific expression of large cargo, such as Cas9, overcoming the limited packaging capacity of a single AAV particle [10].
- Expression Vectors: These are engineered with strong promoters (e.g., CAG, a hybrid of cytomegalovirus enhancer and chicken β-actin promoter) to drive high-level expression of transgenes in neurons [3]. They can also be designed to deliver CRISPR-Cas9 components for targeted genome editing in specific neural cell types [3] [10].
Table 2: Key Vector Types and Their Applications in Neural Studies
| Vector Type | Key Features | Primary Host | Typical Application |
|---|---|---|---|
| Plasmid Vector [7] [1] | Multiple Cloning Site (MCS), Antibiotic Resistance, Origin of Replication (Ori) | E. coli | Gene cloning, protein expression, CRISPR vector construction. |
| AAV Vector [9] [10] | Safe, long-term expression in neurons; cell-type-specific promoters/enhancers; engineered capsids for targeted delivery. | Mammalian cells (in vivo/in vitro) | Gene therapy, functional genomics, neural circuit mapping, in vivo gene editing. |
| Lentiviral Vector [3] | Integrates into host genome for stable expression; can infect non-dividing cells. | Mammalian cells | Creating stable cell lines, expressing shRNA for gene knockdown, delivering large transgenes. |
Host Organisms: The Molecular Factories
The choice of host organism is critical for the cloning and production of recombinant DNA.
- Escherichia coli (E. coli): This bacterium is the most widely used host for plasmid propagation due to its rapid growth and well-understood genetics. Standard competent E. coli strains include NEB 5-alpha and NEB-10 beta [11]. For constructs with repetitive sequences, such as those used in some neural gene studies, specialized strains like NEB Stable are recommended to prevent recombination [11].
- Mammalian Cells: For functional neuroscience studies, the final recombinant DNA is often delivered into mammalian cell lines (e.g., HEK293) or primary neurons to study gene function, protein localization, and signaling pathways in a relevant cellular context [3].
Essential Protocols for Neural Research
Protocol: Traditional Restriction Enzyme Cloning for Plasmid Construction
This protocol is adapted from New England Biolabs (NEB) guidelines [11] and is suitable for constructing plasmids for protein expression or CRISPR guide RNA vectors.
1. Preparation of Insert and Vector:
- Insert Source (PCR Product): Design primers with appropriate restriction sites. Use a proofreading polymerase (e.g., Q5 High-Fidelity DNA Polymerase) for amplification. Purify the PCR product and digest with the chosen restriction enzymes [11].
- Vector: Digest 1 µg of plasmid vector with the same restriction enzymes. To prevent self-ligation, dephosphorylate the vector ends using Antarctic Phosphatase or a similar enzyme [11].
2. Ligation:
- Purify the digested vector and insert.
- Use a molar ratio of 1:3 (vector to insert). For a 4 kb vector and a 1 kb insert, use 50 ng of vector and 37.5 ng of insert [11].
- Assemble the reaction with T4 DNA Ligase or a Quick Ligation Kit. Incubate at room temperature for 5 minutes (quick ligation) or 16°C overnight (standard ligation) [11].
3. Transformation:
- Use recA- strains like NEB 5-alpha Competent E. coli for general cloning.
- Thaw competent cells on ice, add 1-5 µl of the ligation mixture, and incubate on ice for 30 minutes.
- Heat-shock at 42°C for exactly 30 seconds, then place on ice.
- Add recovery medium (e.g., SOC) and incubate at 37°C with shaking for 60 minutes before plating on antibiotic-containing agar plates [11].
4. Screening and Verification:
- Screen colonies by colony PCR or restriction digest of isolated plasmid.
- Verify the final construct by Sanger sequencing, paying close attention to the coding sequence and junctions [7].
Protocol: Utilizing AAV for Cell-Type-Specific Gene Expression in the Mouse Brain
This protocol leverages recent findings on transcriptional crosstalk for delivering large genetic payloads [10].
Objective: To achieve cell-type-specific expression of a large transgene (e.g., Cas9, ~3.2 kb) in a defined neuronal population (e.g., Purkinje cells) after systemic administration.
Workflow:
- Vector Design:
- Enhancer Vector: Package a cell-type-specific enhancer (e.g., Ple155 for cerebellar Purkinje cells) driving a reporter or effector gene into an AAV vector with a BBB-penetrant capsid (e.g., AAV-PHP.eB).
- Cargo Vector: Package your large gene of interest (e.g., Cas9) under a minimal promoter (e.g., minBG, SCP1) into a separate AAV vector with the same tropism.
Virus Production and Purification: Produce high-titer recombinant AAVs using standard methods (e.g., triple transfection in HEK293 cells) and purify via ultracentrifugation or chromatography.
In Vivo Injection:
- Systemically co-inject the two AAVs (Enhancer Vector and Cargo Vector) into wild-type mice via tail vein or retro-orbital injection.
- The AAVs will transduce the target tissue. Upon concatemerization of the AAV genomes in the nucleus, the enhancer on one genome will act in cis on the promoter of the other, driving cell-type-specific expression of the large cargo [10].
Validation:
- Use immunohistochemistry or spatial genomics methods (e.g., AAV-Zombie [10]) to confirm co-localization of the enhancer-driven reporter and the large cargo in the target cell type.
- Assess functional output, such as genome editing efficiency or behavioral phenotypes.
Workflow Visualization
The following diagram illustrates the key steps in a standard restriction enzyme-based cloning workflow, from initial design to verification of the final construct.
Diagram 1: Standard restriction enzyme cloning workflow.
The next diagram outlines the strategy for achieving cell-type-specific expression of large genes using dual AAV vectors and the principle of transcriptional crosstalk.
Diagram 2: Strategy for large cargo delivery via dual AAV crosstalk.
Research Reagent Solutions
Table 3: Essential Materials and Reagents for Molecular Cloning in Neuroscience
| Item Category | Specific Examples | Function & Application Notes |
|---|---|---|
| Restriction Enzymes [11] [8] | EcoRI, BamHI, BsaI (NEB) | DNA cleavage; BsaI for Golden Gate assembly. |
| DNA Ligase [11] [1] | T4 DNA Ligase, Quick Ligation Kit (NEB) | Joins DNA fragments; quick ligation reduces time. |
| Polymerases [11] [1] | Q5 High-Fidelity DNA Polymerase (NEB) | High-fidelity PCR for insert amplification. |
| Competent E. coli [11] | NEB 5-alpha, NEB Stable, NEB 10-beta | Plasmid propagation; NEB Stable for unstable inserts. |
| Cloning Vectors [3] [7] | Plasmid: pUC19, AAV: pAAV, Lentiviral: pLKO.1 | Backbones for gene cloning and delivery. |
| AAV Capsids [9] [10] | AAV9, AAV-PHP.eB, AAV.CAP-B10 | In vivo delivery; engineered for CNS tropism. |
| Cell Lines [3] | HEK293 (for AAV production), Primary Neurons | Protein production and functional assays. |
The development of recombinant DNA technology represents a pivotal revolution in biological science, creating a bridge between fundamental genetic research and transformative clinical applications. This technological paradigm began with the production of recombinant human insulin and has since evolved to enable sophisticated gene delivery systems for the human brain and spinal cord. Molecular cloning, which involves inserting a DNA sequence of interest into an engineered plasmid for propagation within a host organism, laid the foundation for this revolution [12]. The ability to manipulate and express genes across biological systems has not only addressed critical therapeutic shortages but has also opened new frontiers in neuroscience research and the treatment of neurological disorders. This application note details the key historical milestones, experimental protocols, and reagent solutions that have defined the journey from recombinant insulin to neural applications, providing researchers with practical frameworks for advancing this revolutionary technology.
Historical Milestones: From Insulin to Neural Interfaces
The timeline of recombinant DNA technology showcases a rapid progression from conceptual breakthroughs to sophisticated clinical applications. The following table summarizes the key historical milestones that have defined this revolution:
Table 1: Key Historical Milestones in Recombinant DNA Technology
| Year | Milestone | Significance | Key Researchers/Entities |
|---|---|---|---|
| 1922 | Discovery of insulin | First successful pancreatic extract injections for diabetes [13] | Banting, Best, Collip |
| 1972 | First recombinant DNA molecules | Generation of SV40 phages with inserted DNA from lambda phage and E. coli [12] | Paul Berg and colleagues |
| 1973 | Complete restriction enzyme cloning | First execution of sequential digestion, ligation, and transformation [12] | Boyer, Cohen, Chang |
| 1978 | First recombinant human insulin | Preparation of human insulin via recombinant E. coli [13] | David Goeddel (Genentech) |
| 1982 | FDA approval of Humulin | First recombinant pharmaceutical approved for human use [14] | Genentech/Eli Lilly |
| 1983 | Recombinant DNA in neurological disease | Early application of DNA strategies to genetic neurological diseases [15] | Multiple research groups |
| 1987 | Recombinant tPA (Activase) approval | Recombinant enzyme for dissolving blood clots [14] | Genentech |
| 1996 | First short-acting insulin analog | Lispro insulin approved for clinical use [13] | Eli Lilly |
| 2000 | First basal insulin analog | Glargine insulin approved for clinical use [13] | Sanofi |
| 2025 | Neural gene delivery systems | AAV-based systems for targeted brain and spinal cord delivery [16] | NIH BRAIN Initiative |
The initial discovery of insulin in 1922 marked a major breakthrough in medicine, transforming diabetes from a fatal condition to a manageable one [13]. Before insulin, patients with diabetes faced extremely poor prognoses, with children having particularly short life expectancies. The discovery by Banting, Best, and Collip represented the first time a pancreatic extract successfully lowered blood glucose in humans, though early preparations caused sterile abscesses and had variable efficacy [13] [17].
The foundational molecular cloning work in the 1970s established the technical framework for recombinant DNA technology. The discovery of restriction endonucleases—enzymes that site-specifically cut DNA molecules—gave scientists the tools to create the first recombinant DNA molecules [12]. The period from 1972-1973 was particularly significant, with Berg's team creating the first recombinant DNA molecules and Boyer, Cohen, and Chang executing the complete restriction enzyme cloning workflow [12].
The approval of Humulin in 1982 established recombinant DNA technology as a viable industrial process for pharmaceutical production [14]. This first recombinant insulin was produced by inserting genes coding for human insulin into bacteria, which then served as living factories to produce the hormone [14]. The success of Humulin paved the way for numerous other recombinant pharmaceuticals, including growth hormone (Protropin), interferons (Intron A, Roferon-A), and vaccines (Recombivax HB) [14].
Recent advancements have extended these capabilities to neuroscience, with the 2025 development of sophisticated gene delivery systems for targeted brain and spinal cord applications [16]. These systems use adeno-associated viruses (AAVs) to deliver genetic material to specific neural cell types with exceptional accuracy, enabling potential therapies for conditions such as ALS, Parkinson's disease, Alzheimer's disease, and Huntington's disease [16].
Experimental Protocols and Methodologies
Classic Restriction Enzyme-Based Cloning
The foundational protocol for molecular cloning involves several critical steps that remain relevant despite advancements in technology. The following workflow outlines the standard restriction enzyme cloning process:
Diagram 1: Classic restriction enzyme cloning workflow
Step 1: DNA Isolation and Purification
- Procedure: Isolate and purify genomic DNA or PCR-amplified fragments using silica-based column purification methods. For plasmid DNA isolation, use alkaline lysis followed by alcohol precipitation or column-based purification [12].
- Critical Parameters: DNA must be separated from undesired components including other nucleic acids, buffer components, and enzymes from previous steps. Assess DNA purity and concentration using spectrophotometry (A260/A280 ratio of ~1.8) [12].
Step 2: Restriction Enzyme Digestion
- Procedure: Digest both the insert DNA and plasmid vector with the same restriction enzymes that create compatible ends. Use high-fidelity restriction enzymes in optimized buffers, typically incubating at 37°C for 1-2 hours [12].
- Critical Parameters: Include appropriate controls (undigested vector, single digests). Use 2-3 units of enzyme per μg of DNA. For double digests, ensure buffer compatibility or perform sequential digestions with purification steps [12].
Step 3: Ligation
- Procedure: Mix digested insert and vector DNA with T4 DNA Ligase in appropriate buffer. Use a 3:1 molar ratio of insert to vector. Include PEG in the reaction to enhance ligation efficiency. Incubate at 16°C for 4-16 hours [12].
- Critical Parameters: Maintain appropriate vector:insert ratios. Include vector-only controls to assess background. Use high-concentration T4 DNA Ligase for efficient joining of both cohesive and blunt ends [12].
Step 4: Transformation
- Procedure: Introduce ligated DNA into chemically competent or electrocompetent E. coli cells. For chemical transformation, incubate DNA with cells on ice for 30 minutes, heat shock at 42°C for 30-45 seconds, then return to ice. For electroporation, use 1-2 kV for 4-5 ms [12].
- Critical Parameters: Use high-efficiency competent cells (>1×10^8 CFU/μg). Include positive (undigested vector) and negative (no DNA) controls. Allow outgrowth in SOC medium for 1 hour at 37°C with shaking before plating [12].
Step 5: Selection and Screening
- Procedure: Plate transformed cells on agar plates containing appropriate antibiotics. For blue/white screening, include X-gal and IPTG in the plating medium. Incubate at 37°C for 12-16 hours [12].
- Critical Parameters: Screen white colonies for inserts (blue indicates empty vector). Confirm positive clones by colony PCR, restriction analysis, or sequencing [12].
Recombinant Insulin Production Protocol
The production of recombinant human insulin follows a well-established bioprocess with specific parameters for optimal yield:
Fermentation Process:
- Inoculum Preparation: Grow transformed E. coli cells containing proinsulin-producing plasmids in tryptic soy broth with kanamycin monosulfate (0.5g/L) for 24 hours at 37°C [18].
- Bioreactor Parameters: Maintain temperature at 37°C, pH at 7.0, dissolved oxygen at 30% tension. Control foam formation and implement glycerol feeding for continuous carbon source [18].
- Harvesting: Collect cells during late log phase by centrifugation. Resuspend cell pellets in lysis buffer for downstream processing [18].
Purification and Processing:
- Inclusion Body Isolation: Lyse cells using high-pressure homogenization or enzymatic methods. Wash inclusion bodies with detergent solutions to remove membrane components [18].
- Refolding and Cleavage: Solubilize inclusion bodies in denaturing buffer (e.g., 8M urea). Refold proinsulin by gradual removal of denaturant. Cleave proinsulin to insulin using trypsin and carboxypeptidase B [18].
- Final Purification: Use reverse-phase HPLC to purify insulin. Lyophilize final product and formulate with appropriate preservatives (e.g., phenol, cresol) [18].
Neural Gene Delivery System Protocol
Recent advances in gene delivery for neuroscience applications involve sophisticated viral vector systems:
AAV Vector Design and Production:
- Enhancer Selection: Use AI-powered computer programs to identify genetic enhancers that turn genes on in specific brain cell types [16].
- Capsid Selection: Choose AAV serotypes with natural tropism for neural cells or employ engineered capsids for enhanced specificity.
- Vector Packaging: Transfect HEK-293 cells with AAV rep/cap plasmid, adenoviral helper plasmid, and transgene plasmid. Harvest and purify viruses using ultracentrifugation or chromatography methods [16].
Targeted Delivery and Validation:
- Stereotactic Injection: Deliver AAV vectors to specific brain regions using calibrated injection systems. Optimize injection coordinates for target regions (e.g., prefrontal cortex, striatum) [16].
- Dosage Optimization: Titrate viral load based on target region size and desired transduction efficiency. Include fluorescent reporters (e.g., GFP) for visualization.
- Validation Methods: Use immunohistochemistry, in situ hybridization, and electrophysiology to confirm cell-type-specific expression and functional effects [16].
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful implementation of recombinant DNA technologies requires specific reagent systems optimized for each application. The following table details essential research reagents and their functions:
Table 2: Essential Research Reagents for Recombinant DNA Applications
| Reagent Category | Specific Examples | Function and Application | Key Characteristics |
|---|---|---|---|
| Restriction Enzymes | EcoRI, HindIII, BamHI | Site-specific DNA cleavage for fragment generation | High fidelity, optimized buffers, star activity minimization [12] |
| DNA Ligases | T4 DNA Ligase | Joining of DNA fragments with compatible ends | Efficient blunt-end and cohesive-end ligation [12] |
| Cloning Vectors | pBR322, pUC19 | Propagation and maintenance of recombinant DNA | Selectable markers, MCS, replication origins [12] |
| Competent Cells | DH5α, BL21(DE3) | Recombinant DNA uptake and propagation | High transformation efficiency, recA- for stability [12] |
| Expression Systems | E. coli, S. cerevisiae | Recombinant protein production | High yield, proper folding, post-translational modifications [18] |
| Viral Vectors | AAV serotypes (1-9) | Gene delivery to neural cells | Specific tropism, low immunogenicity, long-term expression [16] |
| Purification Systems | Ni-NTA, IgG affinity | Recombinant protein isolation | High specificity, mild elution conditions [18] |
| Selection Agents | Antibiotics, X-gal | Identification of recombinant clones | Clear phenotypic differentiation [12] |
The selection of appropriate host strains is critical for successful molecular cloning. For standard cloning applications, use recA- strains such as DH5α to prevent homologous recombination. For protein expression, employ BL21(DE3) with T7 RNA polymerase systems [12]. For neural gene delivery, select AAV serotypes based on target cell type: AAV1 and AAV2 for broad neural transduction, AAV5 for astrocytes, AAV6 for motor neurons, and AAV9 for blood-brain barrier penetration [16].
Signaling Pathways and Molecular Mechanisms
The biological impact of recombinant DNA technologies operates through specific molecular pathways. The following diagram illustrates the key signaling pathways involved in recombinant insulin action and neural gene delivery:
Diagram 2: Key molecular pathways for insulin signaling and neural gene delivery
Recombinant Insulin Signaling Pathway: Recombinant insulin binds to the insulin receptor, triggering autophosphorylation and activation of its intrinsic tyrosine kinase activity. This leads to phosphorylation of insulin receptor substrate (IRS) proteins, which recruit and activate PI3K. PI3K generates PIP3, recruiting AKT to the membrane where it becomes activated. AKT then promotes GLUT4 translocation to the cell membrane, increasing glucose uptake and regulating metabolic processes [18].
Neural Gene Delivery Pathway: AAV vectors enter target neural cells through receptor-mediated endocytosis. Following internalization, the vector escapes endosomal compartments and translocates to the nucleus where uncoating occurs. The single-stranded DNA genome is converted to double-stranded DNA, enabling transgene transcription. The resulting mRNA is exported to the cytoplasm for translation into therapeutic proteins, which exert their effects through various mechanisms including gene replacement, silencing, or modification of neural circuits [16].
Current Applications and Future Perspectives
Recombinant DNA technology continues to evolve with emerging applications in both metabolic and neurological disorders:
Advanced Insulin Analog Development: Recent research focuses on developing insulin analogs with improved pharmacokinetic profiles. Rapid-acting analogs (lispro, aspart, glulisine) feature amino acid modifications that faster absorption, earlier peak action, and shorter duration [13]. Long-acting analogs (glargine, detemir) employ different strategies: glargine precipitates at injection sites for prolonged absorption, while detemir incorporates a fatty acid chain that binds to albumin, extending its circulation time [13].
Precision Neural Circuit Manipulation: The NIH BRAIN Initiative's "Armamentarium for Precision Brain Cell Access" represents the cutting edge of neural applications. This includes dozens of delivery systems that selectively target key brain cell types, including excitatory neurons, inhibitory interneurons, striatal and cortical subtypes, and hard-to-reach spinal cord neurons [16]. These tools enable researchers to study and potentially treat conditions such as seizure disorders, ALS, Parkinson's disease, Alzheimer's disease, and Huntington's disease [16].
Integration with Artificial Intelligence: AI is playing an increasingly important role in advancing recombinant DNA technologies. AI-powered programs can identify genetic enhancers that turn genes on in specific brain cell types, significantly reducing the time and effort required for this process [16]. In diagnostic applications, AI classifiers have achieved 93% diagnostic accuracy for cancer subtypes, demonstrating the potential for integrating recombinant technologies with computational approaches [19].
The continued evolution of recombinant DNA technology promises to further transform both metabolic disease management and neuroscience research. As these tools become more precise and accessible, they will enable increasingly sophisticated approaches to understanding and treating complex biological systems.
The nervous system represents one of the most complex biological structures in nature, characterized by an exceptional diversity of cell types displaying unique functional connectivity and specialized functions. Recombinant DNA (rDNA) technology has emerged as an indispensable tool for neuroscientists seeking to decipher this complexity by providing precise molecular control over neuronal function. The ability to isolate, modify, and reintroduce genetic material into neural cells has revolutionized our approach to studying brain function, disease mechanisms, and potential therapeutic interventions. rDNA technology comprises altering genetic material outside an organism to obtain enhanced and desired characteristics in living organisms or as their products, involving the insertion of DNA fragments from various sources into vectors containing desirable gene sequences [20].
In the decades since the pioneering recombinant DNA experiments of Paul Berg, Herbert Boyer, and Stanley Cohen in the early 1970s, these methodologies have become fundamentally integrated into neuroscience research [20]. The technology enables neuroscientists to overexpress genes encoding proteins involved in neurodegenerative and neuroprotective events, manipulate neurotransmission pathways, study antioxidant defenses, investigate energetic metabolism, and examine numerous other physiological phenomena related to neuronal homeostasis [21]. This Application Note details the specific methodologies and applications through which rDNA technology addresses the unique challenges presented by nervous system complexity, providing detailed protocols for implementation in neuroscience research settings.
rDNA Toolkit for Neuroscience Research
Essential Vector Systems for Neural Applications
The foundation of rDNA applications in neuroscience rests on specialized vector systems designed to address the unique challenges of working with neuronal cells. These vector systems enable precise genetic manipulation and visualization in complex neural environments.
Table 1: Essential Vector Systems for Neuroscience Applications
| Vector Type | Key Components | Neuroscience Applications | Identification Method |
|---|---|---|---|
| Overexpression Vectors (e.g., pCIG) [21] | Strong promoter (CMV, SV40), MCS, polyadenylation signal, selection marker (Ampr), reporter gene (GFP) [21] | Protein overexpression, dominant-negative mutants, gene function analysis [21] | Fluorescence microscopy (GFP), epitope tags with antibody detection [21] |
| Luciferase Vectors [21] | Regulatory sequence clones, luciferase coding sequence | Study promoter/enhancer activity, transcription factor binding [21] | Luminescence detection (550-570nm) [21] |
| Inducible Promoter Vectors [21] | Signal-responsive promoters (antibiotic/natural molecule) [21] | Temporal control of gene expression, toxic gene studies [21] | Dependent on co-expressed reporter |
| siRNA Vectors [21] | Short hairpin RNA expression cassettes | Gene knockdown studies, functional genomics [21] | Downstream protein detection |
Research Reagent Solutions
Table 2: Essential Research Reagents for Neural rDNA Work
| Reagent/Category | Specific Examples | Function in Neural rDNA Applications |
|---|---|---|
| Cloning Enzymes | Restriction Endonucleases, DNA Ligase [21] [22] | Cut and join DNA fragments for vector construction [21] [22] |
| Bacterial Systems | E. coli K-12 strain [23] | Plasmid propagation and amplification [23] |
| Vector Backbones | Plasmids, Bacteriophages, Cosmids, Artificial Chromosomes [21] | Serve as carriers for genetic material insertion [21] |
| Selection Agents | Ampicillin, Kanamycin, Chloramphenicol [21] | Eliminate non-transformed bacteria [21] |
| Reporter Systems | GFP, Luciferase, Epitope tags (myc, Flag) [21] | Identify transfected cells and protein localization [21] |
| Advanced Engineering Tools | CRISPR-Cas9, Site-directed mutagenesis, DNA assembly methods [22] | Precise genome editing and synthetic biology applications [22] |
Core Methodologies and Protocols
Plasmid DNA Isolation and Purification (Alkaline Lysis Method)
The isolation of plasmid DNA from transformed bacteria represents a fundamental procedure in rDNA workflows. This protocol describes an efficient alkaline lysis method for obtaining high-quality plasmid DNA suitable for transfection of neuronal cultures [21].
Reagents Required:
- LB medium (10% bacterial peptone, 5% yeast extract, 10% NaCl)
- Buffer P1 (Resuspension buffer: 50 mM Tris-HCl pH 8.0, 10 mM EDTA, 100 μg/mL RNAse A)
- Buffer P2 (Lysis buffer: 200 mM NaOH, 1% SDS)
- Buffer P3 (Precipitation buffer: potassium acetate 3M, pH 5.5)
- Cold 100% ethanol
- 80% ethanol
- Sterile water
Procedure:
- Bacterial Culture: Pick a single colony of plasmid-transformed bacteria and inoculate 3 mL of LB medium containing appropriate antibiotic. Grow overnight or until log phase is achieved. Avoid extended growth periods to prevent plasmid loss [21].
- Harvesting: Transfer 1.5 mL of culture to a microcentrifuge tube and centrifuge at 13,000 × g for 20 minutes. Retain remaining culture at 4°C as backup. Pour off supernatant completely [21].
- Resuspension: Add 100 μL of Buffer P1 to the pellet and resuspend vigorously by vortexing until no cell clumps remain [21].
- Lysis: Add 100 μL of Buffer P2. Mix gently by inverting tube 10 times. Do not vortex to prevent genomic DNA contamination. Incubate at room temperature for 3-5 minutes until solution clears [21].
- Neutralization: Add 100 μL of Buffer P3. Mix immediately by inverting 10 times. A white precipitate will form. Centrifuge at 13,000 × g for 5 minutes [21].
- DNA Precipitation: Transfer supernatant to a fresh tube. Add 600 μL of cold 100% ethanol. Mix by inverting 10 times. Centrifuge at 13,000 × g for 5 minutes [21].
- Wash: Pour off supernatant. Add 1 mL of 80% ethanol to pellet. Centrifuge at 13,000 × g for 5 minutes. Pour off supernatant completely [21].
- Resuspension: Air dry pellet for 10-15 minutes. Resuspend in 30 μL sterile water. Store at -20°C [21].
Troubleshooting Notes:
- Low yield may result from incomplete resuspension or short growth time
- RNA contamination indicates RNase A failure - add fresh RNase A to Buffer P1
- Genomic DNA contamination suggests overly vigorous mixing during lysis step
Figure 1: Plasmid DNA Isolation Workflow - This diagram illustrates the step-by-step process for isolating plasmid DNA using the alkaline lysis method, highlighting key reagents and steps from bacterial culture to final DNA preparation.
rDNA Vector Design and Construction for Neuroscience Applications
The design of specialized vectors is crucial for successful neuroscience applications. This protocol details the construction of neural expression vectors optimized for studying gene function in neuronal cultures.
Vector Design Considerations:
- Promoter Selection: Choose promoters based on cell type and expression requirements. The Chicken β-actin promoter combined with CMV immediate-early promoter-enhancer represents an effective choice for cultured neuronal cells [21].
- Reporter Systems: Incorporate GFP or its derivatives for visualization of transfected cells without altering normal cellular homeostasis [21].
- Selection Markers: Include antibiotic resistance genes (Ampr, Kmr, Cmr) for stable transfection selection [21].
- Epitope Tags: Consider adding tags (myc, Flag) for antibody-based detection when fluorescence is not suitable [21].
Restriction Enzyme Cloning Protocol:
- Vector Preparation: Digest 2-5 μg of vector DNA with appropriate restriction enzymes in recommended buffer. Incubate for 2 hours at 37°C [21].
- Insert Preparation: Digest DNA fragment of interest with compatible enzymes. Gel purify fragment if necessary [21].
- Ligation: Combine vector and insert at 3:1 molar ratio with DNA ligase and buffer. Incubate at 16°C for 4-16 hours [21].
- Transformation: Introduce ligation product into competent E. coli via heat shock or electroporation [21].
- Screening: Select colonies on antibiotic plates. Screen for inserts using colony PCR or restriction digest [21].
Advanced Engineering Options:
- Site-Directed Mutagenesis: Create dominant-negative forms of neuronal proteins to study protein function [21] [22]
- CRISPR-Cas9 Systems: Develop knockout models of neuronal genes using guide RNA expression vectors [22]
- Inducible Systems: Implement tetracycline-or antibiotic-responsive promoters for temporal control of gene expression [21]
Advanced Applications in Neuroscience Research
Spatial Transcriptomics and rDNA-Based Cellular Mapping
The brain's complex architecture requires sophisticated mapping approaches to correlate molecular identities with spatial location. Spatial transcriptomics (ST) technologies represent a cutting-edge application of rDNA methodologies that preserve spatial context while enabling comprehensive gene expression analysis [24].
Table 3: Spatial Transcriptomics Technologies in Neuroscience
| Technology | Principle | Resolution | Throughput | Neuroscience Applications |
|---|---|---|---|---|
| MERFISH [24] | Combinatorial barcoding with error-correcting fluorescence in situ hybridization | Single-cell to subcellular | ~10,000 genes | Brain cell type mapping, neural circuit analysis [24] |
| seqFISH/+ [24] | Sequential hybridization with combinatorial barcoding | Subcellular | ~10,000 genes | Brain development, cell type diversity [24] |
| osmFISH [24] | Sequential rounds of single-molecule FISH without barcoding | Single-cell | ~33 transcripts | Detection of low-expression neuronal genes [24] |
| EASI-FISH [24] | Expansion microscopy combined with FISH | Single-cell | Moderate | 3D brain mapping, neuronal connectivity [24] |
| Sequencing-based ST [24] | NGS of captured RNAs from tissue sections | Multicell (50-100μm) | Whole transcriptome | Brain region profiling, disease mapping [24] |
Figure 2: Spatial Transcriptomics Workflow in Neuroscience - This diagram outlines the major approaches for spatial transcriptomics in brain research, comparing imaging-based and sequencing-based methodologies and their applications in mapping neural circuitry and brain function.
rDNA in Neurodegenerative Disease Modeling
Recombinant DNA technology enables precise modeling of neurodegenerative diseases by introducing disease-associated mutations into neuronal cultures and model organisms. These approaches have been instrumental in studying Alzheimer's disease, Parkinson's disease, and amyotrophic lateral sclerosis.
Protocol: Developing Dominant-Negative Mutants for Protein Function Studies
- Target Identification: Select a neuronal protein implicated in disease pathogenesis (e.g., tau, α-synuclein, TDP-43) [21].
- Site-Directed Mutagenesis: Design primers to introduce specific mutations that disrupt normal protein function while maintaining interaction domains [21] [22].
- Vector Construction: Clone mutated sequence into neuronal expression vector with strong promoter and epitope tag [21].
- Validation: Express in heterologous systems to confirm dominant-negative activity before neuronal transfection [21].
- Functional Assays: Assess impact on neuronal viability, synaptic function, and protein aggregation [21].
Recombinant DNA technology continues to be an indispensable component of modern neuroscience research, providing the methodological foundation for addressing the exceptional complexity of the nervous system. From fundamental vector design and plasmid isolation to cutting-edge applications in spatial transcriptomics and neurodegenerative disease modeling, rDNA methodologies enable precise dissection of neural function at molecular, cellular, and circuit levels. The protocols and applications detailed in this Application Note provide neuroscience researchers with practical frameworks for implementing these powerful technologies in their investigations of nervous system function and dysfunction.
As rDNA technologies continue to evolve—with advancements in CRISPR-based genome editing, automated plasmid isolation, and sophisticated expression systems—their integration with neuroscience research promises to yield unprecedented insights into brain function and novel therapeutic approaches for neurological disorders. The ongoing synergy between rDNA methodology development and neuroscience application ensures that these tools will remain essential for addressing the unique challenges presented by the complexity of the nervous system.
From Sequence to Synapse: Practical Cloning Methods and Their Groundbreaking Neuroscience Applications
A Guide to PCR-Based Cloning for Cellular Engineering in Neural Models
Molecular cloning is a foundational pillar of biological research, enabling the study and manipulation of genetic material. Within neuroscience, the ability to precisely engineer neural cell models is crucial for dissecting the molecular mechanisms of brain function, neuronal development, and neurological diseases. PCR-based cloning represents a powerful and versatile method for the rapid construction of recombinant DNA, offering significant advantages for projects that require higher throughput than traditional cloning methods can accommodate [25]. This application note details the implementation of PCR-based cloning, providing a structured protocol and resource guide framed within the context of cellular engineering for neural models. This technique allows for the cloning of DNA fragments that are not available in large amounts, making it particularly suitable for working with precious neuronal cDNA or low-abundance transcripts [25].
Core Principles and Comparative Analysis
At its simplest, PCR-based cloning involves amplifying a gene of interest (GOI) using polymerase chain reaction (PCR) and, in the process, adding necessary sequences to its ends to facilitate its insertion into a plasmid vector [26]. This method diverges from traditional restriction enzyme cloning by using PCR, rather than restriction enzymes alone, to generate the insert, providing greater flexibility in vector choice and insert design [27] [25].
The table below provides a technical comparison of PCR cloning with other common gene cloning methods, highlighting key considerations for experimental planning in neural research.
Table 1: Comparative Analysis of Common Cloning Methods
| Method | Mechanism | Key Applications | Key Advantages | Key Limitations |
|---|---|---|---|---|
| PCR Cloning [25] [26] | Amplification of insert (and/or vector) via PCR; ligation via sticky/blunt ends or dedicated kits. | High-throughput cloning, expression constructs, cellular engineering. | Rapid; high efficiency with dedicated vectors; amenable to high throughput; doesn't require large amounts of source DNA. | Limited vector choices can be expensive; difficult directional and multi-fragment cloning; higher risk of mutation. |
| Traditional Restriction Cloning [27] | Digestion of insert and vector with restriction enzymes; ligation. | Generation of expression constructs, library construction. | Robust, widely taught and understood; highly standardized. | Dependent on availability of restriction sites; can be labor-intensive; lower throughput. |
| Golden Gate Assembly [27] | Uses Type IIS restriction enzymes to create user-defined overhangs; simultaneous digestion and ligation. | Complex pathway engineering, synthetic gene circuits, modular assembly. | Seamless, directional, and scarless; can assemble many fragments in a single reaction. | Requires careful sequence design to avoid internal enzyme sites; sensitive to sequence context. |
A critical design consideration for any cloning project is the selection of restriction enzymes. For PCR cloning, these sites are incorporated into the PCR primers. Ideal enzymes should be single cutters within the vector, generate sticky ends for higher efficiency, and not be sensitive to DNA methylation from standard E. coli strains used for plasmid propagation [28]. Furthermore, using enzymes that are active in the same buffer allows for a simultaneous double digest, saving time and reducing DNA loss [28].
Experimental Workflow and Visualization
The following diagram illustrates the logical flow of a standard PCR-based cloning experiment, from initial design to verification of the final plasmid.
Detailed Experimental Protocol
Primer Design and PCR Amplification
The success of PCR cloning hinges on meticulous primer design [26] [28]. Primers must include sequences for both specific hybridization to the template and for subsequent cloning steps.
Design Principles: A standard cloning primer is composed of three parts:
- 5' Leader Sequence (3-6 bp): Extra bases (e.g.,
TAAGCA) added to ensure efficient restriction enzyme cleavage at the ends of the PCR product [26]. - Restriction Site (6-8 bp): The recognition sequence for the chosen restriction enzymes (e.g.,
GAATTCfor EcoRI). The forward primer incorporates the upstream site, and the reverse primer incorporates the downstream site [26]. - Hybridization Sequence (18-21 bp): The region that binds specifically to the gene of interest to be amplified. The annealing temperature is calculated based on this portion of the primer [26].
- 5' Leader Sequence (3-6 bp): Extra bases (e.g.,
Performing PCR: Amplify your gene of interest using a high-fidelity DNA polymerase to minimize the introduction of mutations during amplification [26]. The fidelity of the polymerase is especially critical for longer genes. The annealing temperature should be optimized based on the Tm of the hybridization sequence, not the entire primer [26].
Digestion, Ligation, and Transformation
Purification and Digestion: Purify the PCR product using a commercial kit [26]. Set up restriction digests for both the purified PCR product and the recipient plasmid. It is critical to achieve complete digestion; therefore, digest for at least 4 hours or overnight [26]. To prevent vector self-ligation, the linearized vector can be treated with a phosphatase (e.g., CIP or SAP) [26] [28].
Ligation: Isolate the digested insert and vector fragments by gel purification [26]. For the ligation reaction, a molar ratio of approximately 1:3 (vector to insert) is often effective. It is crucial to include a negative control (vector alone) to assess background from uncut or self-ligated vector [26].
Transformation and Screening: Transform 1-2 µl of the ligation reaction into competent E. coli cells, such as DH5α [26]. Screen resulting colonies by colony PCR or analytical restriction digest of purified plasmid DNA. Finally, verify the plasmid by sequencing the entire insert, as PCR-based cloning carries a higher risk of mutation than traditional methods [26].
The Scientist's Toolkit: Essential Reagents
Table 2: Key Research Reagents for PCR-Based Cloning
| Reagent / Material | Function / Explanation | Examples / Notes |
|---|---|---|
| High-Fidelity DNA Polymerase | Amplifies the gene of interest with minimal error rates. | Essential for accurate gene representation; error rates can range from ~1/500bp and lower [26]. |
| Restriction Endonucleases | Enzymes that cut DNA at specific sequences to create ends for ligation. | Choose enzymes that are not sensitive to Dam/Dcm methylation and function in a single buffer [28]. |
| T4 DNA Ligase | Enzyme that covalently joins compatible ends of DNA fragments. | Used to ligate the digested PCR insert into the prepared vector backbone [27]. |
| Cloning Vector | Plasmid backbone for propagating and expressing the insert. | Vectors with toxic "suicide genes" can improve selection efficiency [25]. |
| Competent E. coli Cells | Bacterial cells rendered permeable for DNA uptake. | Standard strains like DH5α are sufficient for most cloning; high-efficiency cells are for low DNA amounts [26] [28]. |
| Gel Purification Kit | Isolates DNA fragments of the correct size from an agarose gel. | Critical for removing undigested plasmid or incorrect fragments before ligation [26]. |
Applications in Neural Model Engineering
PCR-based cloning is incredibly versatile for neural research. It is ideally suited for tasks such as:
- Constructing Expression Vectors: Copying a neuronal cDNA (e.g., for a ion channel, receptor, or synaptic protein) from one vector into a new vector better suited for functional analysis [26].
- Cellular Engineering: As demonstrated in a 2015 protocol, PCR cloning can be used to generate fluorescent protein-expressing constructs (e.g., tdTomato) for transduction into target cells, enabling cell tracking and visualization [28]. This is directly applicable to creating engineered neural cell lines for in vitro and in vivo studies.
- Chimeric and Mutagenic Constructs: Advanced PCR methods like FastCloning leverage overlapping primers and DpnI digestion to seamlessly integrate inserts, making it ideal for constructing fusion proteins (e.g., GFP-tagged proteins) or introducing specific mutations without relying on restriction sites [29].
Quantitative Data and Efficiency Analysis
The table below summarizes key quantitative metrics related to cloning efficiency and analysis, derived from the literature.
Table 3: Quantitative Data on Cloning and Analysis Methods
| Parameter | Method / Context | Reported Value / Finding | Implication |
|---|---|---|---|
| PCR Error Rate | PCR amplification for cloning [26] | ~1 error per 500 base pairs (range provided) | Necessitates sequencing of the final construct. |
| qPCR Data Analysis Precision | Weighted Linear Regression vs Simple Linear Regression [5] | Coefficient of Variation (CV) reduced from ~25.4% (SLR) to ~18.3% (WLR) | Weighted models improve precision in quantitative analysis. |
| Direct Ligation Efficiency | Annealing-free short DNA fragment cloning [30] | >80% positive colonies with paired oligos. | Bypassing annealing and PCR is efficient for short inserts. |
| Positive Colony Yield | Cloning with low self-ligation ends (NcoI/SalI) [30] | ~90% positive colonies. | Restriction site choice drastically impacts screening workload. |
Molecular cloning is a foundational technique in molecular biology, enabling researchers to create recombinant DNA molecules for a wide array of applications. In neuroscience research, these methods facilitate the study of neuronal gene expression, protein function, and cellular signaling pathways, ultimately advancing our understanding of brain function and neurological disorders. The selection of an appropriate cloning strategy is paramount to experimental efficiency and success. This application note provides a detailed comparison of three widely used techniques—Restriction Enzyme Cloning, Gibson Assembly, and Gateway Cloning—to guide researchers in selecting the optimal method for their specific research context.
Restriction Enzyme Cloning
Restriction Enzyme Cloning, also referred to as subcloning, is one of the earliest developed cloning methods. It relies on the use of restriction endonucleases—enzymes that recognize and cleave specific DNA sequences—to generate compatible ends on both the insert and vector DNA fragments. These fragments are then joined together by DNA ligase to form a recombinant plasmid [3] [31]. This method was pioneered following the discovery of Type II restriction enzymes, a breakthrough that earned Werner Arber, Hamilton Smith, and Daniel Nathans the 1978 Nobel Prize [3].
Detailed Experimental Protocol
The following protocol outlines the key steps for subcloning a DNA fragment from a donor plasmid into a recipient vector [31].
Step 1: Experimental Design and Restriction Enzyme Selection Identify restriction enzymes that:
- Flank your insert but do not cut within it.
- Are present in the Multiple Cloning Site (MCS) of your recipient plasmid.
- Will result in your insert being in the correct orientation. Ideally, use two different enzymes to prevent vector re-circularization.
Step 2: Restriction Digest Set up separate digestions for your donor plasmid (1.5-2 µg) and recipient plasmid (1 µg). For the recipient plasmid, a digestion time of 4 hours to overnight is critical to ensure complete cutting. If using a single enzyme or enzymes with compatible ends, treat the digested recipient plasmid with a phosphatase (e.g., CIP or SAP) to prevent self-ligation.
Step 3: Gel Purification Run the digested DNA on an agarose gel. Excise the bands corresponding to the linearized vector backbone and your insert. Purify the DNA fragments from the gel using a gel extraction kit and determine their concentrations.
Step 4: Ligation Ligate the purified insert and vector backbone using T4 DNA ligase. A typical reaction uses ~100 ng of total DNA with a vector-to-insert molar ratio of 1:3. Incubate at room temperature for 10-30 minutes. Always include a negative control (vector alone) to assess background ligation.
Step 5: Transformation and Screening Transform 1-2 µL of the ligation reaction into competent E. coli cells (e.g., DH5α). Plate the cells on selective media and incubate overnight. The following day, pick several colonies, culture them, and purify the plasmid DNA. Verify successful cloning via a diagnostic restriction digest, which should yield two bands: one for the vector and one for the insert [31].
Research Reagent Solutions
| Reagent | Function |
|---|---|
| Restriction Endonucleases (e.g., EcoRI, HindIII) | Enzymes that recognize and cleave specific DNA sequences to generate compatible ends. |
| T4 DNA Ligase | Enzyme that catalyzes the formation of phosphodiester bonds between adjacent nucleotides, joining DNA fragments. |
| DNA Polymerase (High-Fidelity) | Used in PCR to amplify the insert and potentially add restriction sites if they are not present. |
| Agarose Gel Electrophoresis System | Used to separate and visualize DNA fragments by size after restriction digest. |
| Chemically Competent E. coli | Bacterial cells treated to readily take up foreign DNA during transformation. |
| Plasmid Miniprep Kit | For isolating and purifying plasmid DNA from bacterial cultures for screening. |
Gibson Assembly
Gibson Assembly is an advanced, seamless cloning method that allows for the in vitro assembly of multiple overlapping DNA fragments in a single, isothermal reaction. Developed by Daniel Gibson in 2009, this technique employs a master mix containing three enzymes that work in concert: a 5' exonuclease chews back DNA ends to create single-stranded overhangs; a DNA polymerase fills in the gaps; and a DNA ligase seals the nicks in the DNA backbone [32] [33]. Its flexibility and efficiency make it ideal for assembling large constructs, such as entire viral genomes for vaccine development or complex gene circuits for neuroscience applications [32].
Detailed Experimental Protocol
Step 1: Fragment Preparation with Homology Arms Amplify the DNA fragments to be assembled (insert and linearized vector) by PCR. The primers must be designed to add 20-40 base pair overlapping homologous sequences to the ends of each fragment. These overlaps are critical for the correct assembly of the fragments.
Step 2: Gibson Assembly Reaction Combine the linearized vector and insert(s) with the Gibson Assembly master mix. A typical reaction might use 100-200 ng of total DNA. There is no need for gel purification of the PCR products if a high-fidelity polymerase was used.
Step 3: Incubation Incubate the reaction at 50°C for 30-60 minutes. The isothermal conditions allow all three enzymes to function simultaneously.
Step 4: Transformation and Screening Transform 1-2 µL of the assembly reaction directly into competent E. coli cells. Screen colonies by colony PCR or diagnostic restriction digest to confirm correct assembly [32] [33].
Research Reagent Solutions
| Reagent | Function |
|---|---|
| Gibson Assembly Master Mix | A proprietary blend containing T5 exonuclease, DNA polymerase (e.g., Phusion), and DNA ligase. |
| High-Fidelity DNA Polymerase | For error-free PCR amplification of DNA fragments with added homologous overlaps. |
| Chemically Competent E. coli | For transformation of the assembled plasmid. |
| DNA Purification Kit | For cleaning up PCR products prior to assembly (optional, depending on protocol). |
Gateway Cloning
Gateway Cloning is a versatile, site-specific recombinational cloning system developed by Invitrogen. It is based on the bacteriophage λ integration and excision system, which utilizes specific attachment (att) sites and enzyme mixes (BP Clonase and LR Clonase) to shuttle DNA sequences between vectors in a highly efficient and standardized manner [34] [35]. This method is exceptionally well-suited for high-throughput applications, such as transferring a library of neuronal genes into various expression vectors for functional screening.
Detailed Experimental Protocol
The Gateway system involves two primary recombination reactions [34].
BP Reaction: Creating an Entry Clone
- Amplify your gene of interest (GOI) using primers that add attB sites to its ends.
- Set up the BP reaction by mixing the attB-flanked PCR product, a Donor vector (containing attP sites), and BP Clonase II enzyme mix.
- Incubate at 25°C for 1 hour.
- Terminate the reaction with Proteinase K and transform into competent cells. The resulting plasmid is an "Entry Clone," which contains the GOI flanked by attL sites.
LR Reaction: Creating an Expression Clone
- Mix the Entry Clone (containing your GOI), a Destination Vector (containing attR sites and the desired promoter/reporter for neuronal expression), and LR Clonase II enzyme mix.
- Incubate at 25°C for 1 hour.
- Terminate with Proteinase K and transform into competent cells. The resulting "Expression Clone" contains the GOI now transferred into the Destination Vector [34].
A "One-Tube" format is also available, which combines the BP and LR reactions to create an expression clone directly from a PCR product [34].
Research Reagent Solutions
| Reagent | Function |
|---|---|
| Donor Vector (e.g., pDONR) | Contains attP sites and a ccdB//CmR cassette for selection during the BP reaction. |
| Destination Vector | Contains attR sites and a promoter/reporter system; the gene is inserted in place of the ccdB gene. |
| BP Clonase II Enzyme Mix | Enzyme cocktail that catalyzes recombination between attB and attP sites. |
| LR Clonase II Enzyme Mix | Enzyme cocktail that catalyzes recombination between attL and attR sites. |
| ccdB-Sensitive Competent Cells (e.g., DH5α) | For transformation of LR reactions; only cells with recombined plasmid (lacking ccdB) survive. |
| DB3.1 Competent E. coli | A ccdB-resistant strain used for propagating Gateway vectors containing the toxic ccdB gene. |
Comparative Analysis
The table below provides a direct comparison of the key characteristics of the three cloning methods to aid in selection.
Table 1: Comparative Analysis of Cloning Methods
| Feature | Restriction Enzyme Cloning | Gibson Assembly | Gateway Cloning |
|---|---|---|---|
| Principle | Restriction digestion & ligation [31] | Homologous recombination in vitro [33] | Site-specific recombination (att sites) [35] |
| Seamlessness | Leaves scar sequences [3] | Seamless (scarless) [33] | Seamless (scarless) [35] |
| Typical Fragment Capacity | 1-2 fragments | Up to ~15 fragments [33] | 1 fragment per reaction (multisite available) [35] |
| Key Requirement | Compatible restriction sites absent from the insert [31] | 20-40 bp homologous overlaps [33] | Specific att sites on vectors and inserts [34] |
| Efficiency | Moderate | High [33] | Very High [35] |
| Cost | Low | Generally more expensive [33] | Expensive (commercial vectors/enzymes) [3] [35] |
| Throughput | Low | Moderate | High (ideal for 96-well format) [35] |
| Best For | Simple subcloning, when restriction sites are available and convenient. | Assembling multiple fragments, large constructs, and seamless mutagenesis. [33] [36] | High-throughput transfer of genes into multiple expression systems. [35] |
Workflow Diagrams
The choice of a cloning method is a strategic decision that depends on the experimental goals, available resources, and required throughput.
- Restriction Enzyme Cloning is a reliable and cost-effective choice for straightforward subcloning tasks where suitable restriction sites are available and not present within the gene of interest. Its simplicity makes it a good entry point for many labs.
- Gibson Assembly excels in flexibility and is the method of choice for complex projects involving the assembly of multiple DNA fragments, the construction of large vectors (e.g., for viral packaging in neuronal tracing studies), or when the introduction of specific mutations without extra nucleotides is required [32] [36].
- Gateway Cloning is unparalleled in high-throughput environments. Its standardized system is ideal for transferring a single gene into dozens of different expression vectors (e.g., for neuronal cell-type-specific expression or protein localization studies) or for managing large collections of genes, such as an ORFeome library [35].
For neuroscience research specifically, Gibson Assembly is highly valuable for building complex constructs for optogenetics, chemogenetics (DREADDs), or CRISPR-Cas9 gene editing applications. Gateway Cloning streamlines the process of testing a gene's function across multiple cellular models (e.g., primary neurons, astrocyte cultures, and in vivo). By understanding the strengths and applications of each method, researchers can optimize their molecular cloning strategies to accelerate discovery in the neurosciences.
The study of neural proteins, such as receptors, ion channels, and signaling molecules, is fundamental to advancing our understanding of brain function and neurological disorders. Heterologous expression of these proteins in recombinant systems is a cornerstone of neuroscience research, enabling structural studies, drug screening, and functional characterization. However, achieving high-yield expression of functional neural proteins presents significant challenges, as the native coding sequences of neural genes are often poorly expressed in standard expression hosts like Escherichia coli, yeast, or mammalian cell lines [37] [38].
This application note details a structured approach to gene design, focusing on state-of-the-art codon optimization techniques integrated within the broader context of molecular cloning and recombinant DNA technology. We provide neuroscience researchers with actionable protocols and data-driven strategies to overcome translational barriers, thereby enhancing the production of high-quality neural proteins for downstream applications.
The Scientific Basis of Codon Optimization
Genetic Code Degeneracy and Codon Usage Bias
The genetic code is degenerate, meaning most amino acids are encoded by multiple nucleotide triplets, or codons. Organisms exhibit a non-random preference for certain synonymous codons, a phenomenon known as codon usage bias [38]. This bias reflects a balance between mutational pressures and natural selection for translational optimization and is a species-specific characteristic [39] [37]. For example, the codons TCT and GCT are more frequent in highly expressed E. coli genes, whereas TTA and ATA are rare [39].
The primary consequence of this bias is that the abundance of transfer RNA (tRNA) molecules, which deliver amino acids to the ribosome, correlates with the frequency of their cognate codons in highly expressed genes [40] [38]. When a heterologous gene, such as one of human neural origin, is expressed in a host like E. coli, it may contain a high frequency of codons that correspond to low-abundance tRNAs in the host. This mismatch can lead to ribosomal stalling, translation errors, premature termination, and reduced protein yields [37] [38]. Furthermore, the rate of translation influenced by codon choice can impact the correct co-translational folding of the nascent protein, which is critical for the function of complex neural proteins [39] [38].
Key Parameters for Effective Optimization
Modern codon optimization moves beyond simple rare-codon elimination. It is a multi-parameter process that harmonizes various sequence features to maximize transcriptional and translational efficiency while ensuring proper protein folding [41] [37]. Key parameters include:
- Codon Adaptation Index (CAI): This is a quantitative measure of how similar a gene's codon usage is to the preferred codon usage of a reference set of highly expressed genes in the target host. A CAI value closer to 1.0 indicates a stronger alignment with host bias and is generally predictive of higher expression levels [41] [42] [43].
- tRNA Adaptation Index (tAI): This index predicts expression levels based on the pool of available tRNAs and their binding efficiencies, offering a more direct measure of translational capacity than CAI [39].
- GC Content: The percentage of guanine and cytosine nucleotides in a sequence significantly impacts mRNA stability and secondary structure. Optimal GC content is host-specific; for instance, high GC content can stabilize mRNA in E. coli but may create overly stable secondary structures that impede translation initiation in other systems [41].
- mRNA Secondary Structure: Stable secondary structures, particularly in the 5' end near the translation initiation site, can dramatically reduce protein expression by blocking ribosome binding and scanning [37]. The stability of these structures is often assessed by calculating the minimum folding energy (ΔG).
- Codon Context and CpG Dinucleotides: The specific pairing of adjacent codons (codon-pair bias) can influence translation efficiency [41]. Additionally, CpG dinucleotides can be targets for methylation, which may affect gene expression in mammalian systems and should be considered in their design [37].
- Cryptic Splice Sites and Regulatory Motifs: The optimized sequence must be scanned and modified to avoid unintended sequence motifs, such as cryptic splicing signals in mammalian cells, internal ribosome entry sites, RNA instability motifs (ARE), and restriction enzyme sites used in subsequent cloning steps [44] [37].
Table 1: Key Parameters for Codon Optimization and Their Impact on Protein Expression.
| Parameter | Description | Impact on Expression | Optimal Range (Host-Dependent) |
|---|---|---|---|
| Codon Adaptation Index (CAI) | Measures similarity to host's highly expressed genes | Higher CAI (≥0.8) correlates with higher translation efficiency [42] | 0.8 - 1.0 |
| GC Content | Percentage of Guanine and Cytosine bases | Affects mRNA stability and secondary structure; extremes can hinder transcription/translation [41] [42] | ~50-60% (varies by host) |
| mRNA Secondary Structure (ΔG) | Stability of intramolecular base-pairing, especially at 5' end | Stable 5' structures can block ribosome access, reducing yield [37] | Minimize stability at 5' UTR |
| Codon-Pair Bias (CPB) | Frequency of specific adjacent codon pairs | Can influence translation speed and accuracy [41] | Match host genome bias |
| tRNA Adaptation Index (tAI) | Measures compatibility with host's tRNA pool | Better adaptation leads to faster, more accurate translation [39] | Higher is better |
A Practical Workflow for Optimizing Neural Protein Expression
The following integrated workflow outlines the key steps from gene design to experimental validation, specifically tailored for neural proteins.
Diagram 1: A workflow for recombinant neural protein production.
Protocol: Implementing a Multi-Tool Optimization Strategy
Different optimization algorithms employ distinct strategies, leading to varied sequence outputs and potential differences in experimental outcomes. A comparative approach is recommended [41].
Materials:
- Amino acid sequence of the target neural protein (e.g., in FASTA format).
- Computer with internet access.
- List of preferred expression hosts (e.g., E. coli BL21, HEK293, Sf9).
Method:
- Sequence Submission: Submit your target protein sequence to several codon optimization tools. Recommended tools include:
- JCAT: For its strong alignment with host codon usage and simple interface [41].
- OPTIMIZER or ATGme: For their ability to use reference sets of highly expressed genes [41].
- IDT Codon Optimization Tool or VectorBuilder's Tool: For their user-friendliness and integration with gene synthesis services [45] [42].
- TISIGNER: For its focus on optimizing the 5' end to avoid stable mRNA secondary structures [41].
- Parameter Setting: For each tool, select your target expression host (e.g., E. coli K12, H. sapiens). Use default parameters for an initial comparison.
- Sequence Retrieval: Download the resulting DNA sequences in FASTA or GenBank format.
Protocol: Analytical Comparison of Optimized Sequences
Once multiple optimized sequences are obtained, a comparative analysis is crucial for selecting the best candidate for synthesis.
Materials:
- Optimized DNA sequences from Step 3.1.
- Bioinformatics tools for sequence analysis (e.g., VectorBuilder's tool suite, GenScript's resources [44] [42]).
Method:
- Calculate Key Metrics: For each optimized sequence, calculate the following:
- CAI: Use an online CAI calculator, specifying the host organism.
- GC Content: Calculate the overall GC percentage.
- Codon Frequency Distribution: Compare the usage of each codon for an amino acid to the host's ideal frequency table [44].
- Check for Undesirable Motifs: Scan sequences for the accidental introduction of restriction sites, direct repeats, or RNA instability motifs (AREs) [37].
- Rank Sequences: Create a decision matrix, like the one below, to rank the sequences based on a balanced assessment of all parameters. Do not select on CAI alone.
Table 2: Comparative Analysis of Sequences Optimized by Different Tools for a Hypothetical Neural Receptor Expressed in E. coli.
| Optimization Tool | CAI | GC Content | 5' ΔG (kcal/mol) | Notes |
|---|---|---|---|---|
| JCAT | 0.95 | 52% | -5.2 | Excellent CAI, moderate GC, unstable 5' end |
| OPTIMIZER | 0.91 | 48% | -8.5 | Good CAI, low GC, more stable 5' structure |
| IDT | 0.89 | 55% | -4.1 | Lower CAI, higher GC, very unstable 5' end |
| Native Sequence | 0.65 | 45% | -12.3 | Poor CAI, contains multiple rare codons |
Experimental Validation and Troubleshooting
Protocol: Small-Scale Expression Test and Analysis
After selecting and synthesizing the top candidate gene(s), cloning them into an appropriate expression vector, and transforming into the host organism, protein expression must be empirically validated.
Materials:
- Transformed expression host (e.g., E. coli BL21(DE3) with plasmid).
- Appropriate culture media and inducers (e.g., LB broth, IPTG).
- Lysis buffer (e.g., containing lysozyme for bacteria, detergents for mammalian cells).
- SDS-PAGE gel equipment.
- Western blot supplies (if antibodies are available).
- Reagents for functional assays (e.g., binding, enzymatic activity).
Method:
- Induction Test: Inoculate a small culture (5-10 mL) and grow to mid-log phase. Induce protein expression (e.g., with IPTG) and continue incubation for an optimized duration and temperature.
- Harvesting and Lysis: Pellet the cells by centrifugation. Resuspend the pellet in lysis buffer and disrupt the cells by sonication (for bacteria) or detergent treatment.
- Fractionation: Centrifuge the lysate at high speed to separate the soluble (supernatant) and insoluble (pellet) fractions.
- SDS-PAGE Analysis: Load equal proportions of total lysate, soluble fraction, and insoluble fraction onto an SDS-PAGE gel. Compare the bands to an uninduced control to identify the protein band of the correct molecular weight.
- Functional Validation: For the soluble protein, perform a quick activity assay. For a receptor, this could be a ligand-binding assay; for an enzyme, a specific activity measurement. This step is critical to confirm that the optimized protein is not only highly expressed but also functionally folded.
The Scientist's Toolkit: Essential Reagents for Expression
Table 3: Key Research Reagent Solutions for Recombinant Neural Protein Production.
| Reagent / Material | Function / Application | Example / Notes |
|---|---|---|
| Codon-Optimized Gene | Template for transcription/translation; maximizes compatibility with host machinery. | Synthesized de novo based on in silico design [40]. |
| Expression Vectors | Plasmid DNA carrying regulatory elements (promoter, RBS) for controlled gene expression. | pET vectors (for E. coli), pCEP4 (for HEK293 cells), pPICZ (for yeast). |
| Specialized Host Strains | Expression hosts engineered to overcome specific limitations. | Rosetta E. coli: Supplies rare tRNAs [38]. HEK293: For post-translational modifications. |
| Lysis Reagents | Disrupt cellular membranes to release the recombinant protein. | Lysozyme (bacteria), Detergents (e.g., Triton X-100), Protease inhibitor cocktails. |
| Affinity Chromatography Resins | Purify the recombinant protein based on a fused tag. | Ni-NTA Resin (for His-tagged proteins), Glutathione Sepharose (for GST-tagged proteins). |
The successful high-yield expression of neural proteins is an achievable goal through the rational application of gene synthesis and multi-parameter codon optimization. By moving beyond simplistic rare-codon replacement and adopting a holistic strategy that considers mRNA structure, host-specific biases, and protein folding, neuroscience researchers can significantly enhance their experimental outcomes. The protocols and analytical frameworks provided herein serve as a comprehensive guide for designing genes that are not just optimized in silico, but are proven to be highly expressive and functional at the bench, thereby accelerating research in molecular neuroscience and drug discovery.
Recombinant DNA (rDNA) technology serves as the foundational engine for modern optogenetics, a field that has revolutionized neuroscience by enabling precise, millisecond-scale control over specific neural populations. By allowing scientists to genetically embed light-sensitive ion channels, or opsins, into targeted neurons, optogenetics facilitates the direct interrogation of complex brain circuits. The development and application of advanced optogenetic tools like Chronos and Chrimson are quintessential achievements of molecular cloning. These opsins, discovered through large-scale genomic screening and optimized via rational protein engineering, offer unprecedented capabilities for high-temporal-precision neural activation and multi-color experiments. This Application Note details the practical application of rDNA technology to harness these powerful tools, providing structured protocols, quantitative comparisons, and essential resources for researchers aiming to dissect neural circuitry with light.
The Optogenetic Toolbox: Chronos and Chrimson
The expansion of the optogenetic arsenal through recombinant DNA methods has been critical for addressing diverse experimental needs. Among the most powerful tools are the channelrhodopsins Chronos and Chrimson, which were identified through the de novo transcriptome sequencing of over 100 species of algae [46].
Key Optogenetic Actuators
- Chronos: A channelrhodopsin from Stigeoclonium helveticum, nicknamed Chronos for its rapid kinetics. It is a blue-green light-drivable opsin (peak action spectrum ~500 nm) with the fastest turn-on (2.3 ± 0.3 ms) and turn-off (3.6 ± 0.2 ms) kinetics reported at the time of its discovery, enabling reliable neural spiking at frequencies up to 60 Hz [46].
- Chrimson: A channelrhodopsin from Chlamydomonas noctigama, is the most red-shifted channelrhodopsin known (peak action spectrum ~590 nm), which is 45 nm more red-shifted than any prior tool [46]. Its initial slow off-kinetics (tau-off ~21.4 ms) were improved via site-directed mutagenesis to create ChrimsonR (K176R mutation), which has a faster turn-off (15.8 ± 0.4 ms) and supports reliable spiking at up to 20 Hz [46].
The distinct spectral and kinetic profiles of these tools make them ideal for independent two-color activation of distinct neural populations in the same brain tissue, a previously unattainable goal [46].
Quantitative Comparison of Microbial Opsins
Table 1: Characteristics of Key Optogenetic Actuators
| Opsin Name | Type | Peak Action Spectrum (nm) | Kinetics (Tau-off) | Key Feature(s) | Primary Experimental Use |
|---|---|---|---|---|---|
| ChR2 | Cation Channel | ~470 [47] | ~10 ms [48] | Foundational tool | General neural excitation |
| Chronos | Cation Channel | ~500 [46] | ~3.6 ms [46] | Very fast kinetics, high light sensitivity | High-frequency neural stimulation |
| Chrimson | Cation Channel | ~590 [46] [49] | ~21.4 ms [46] | Most red-shifted peak | Deep tissue penetration; paired with blue-light tools |
| ChrimsonR | Cation Channel | ~590 [46] | ~15.8 ms [46] | Red-shifted peak, faster kinetics than WT | Improved temporal precision with red light |
| Halo/NpHR | Chloride Pump | ~589 [47] | Slow (tens of ms) | Neural inhibition | Silencing neuronal activity |
| Arch | Proton Pump | ~566 [47] | Fast (ms) | Neural inhibition | Silencing neuronal activity |
Molecular Cloning Strategies for Optogenetic Constructs
The delivery of opsin genes into neurons relies on robust molecular cloning techniques to create plasmid vectors or viral vectors (e.g., Adeno-Associated Virus, AAV) for expression. The choice of cloning method depends on the experimental requirements for speed, efficiency, and complexity.
Cloning Techniques and Workflow
A standard workflow involves isolating the opsin gene, inserting it into a plasmid vector under a cell-type-specific promoter, and packaging it into viral particles for delivery. Common techniques include:
- Restriction Enzyme Cloning: A classical method using restriction enzymes to cleave DNA at specific sites and DNA ligase to insert the opsin gene into a plasmid vector [50] [51]. Although reliable, it can be time-consuming.
- Gateway Cloning: A recombination-based system that allows for the efficient transfer of DNA sequences between different vector platforms, which is ideal for high-throughput applications where the same opsin needs to be expressed from different promoters [51].
- Gibson Assembly: An advanced, seamless method that uses a combination of exonuclease, polymerase, and ligase to assemble multiple DNA fragments in a single reaction. This is particularly useful for building complex constructs, such as those combining an opsin with a fluorescent protein and a specific promoter [51].
The following diagram illustrates a generalized recombinant DNA cloning workflow for creating an optogenetic construct.
Experimental Protocol: Classical Restriction Enzyme Cloning of an Opsin Gene
This protocol outlines the key steps for inserting an opsin gene into a plasmid vector using restriction enzyme digestion and ligation [50].
Step 1: Amplify Gene of Interest
- Design PCR primers that incorporate specific restriction enzyme sites (e.g., EcoRI, NotI) at the 5' and 3' ends of the opsin gene (e.g., ChrimsonR).
- Perform PCR amplification to generate a sufficient quantity of the insert.
Step 2: Digest Insert and Vector
- Purify the PCR product.
- Set up parallel digestion reactions for both the purified insert and the plasmid vector using the selected restriction enzymes.
- Incubate according to the enzyme manufacturer's specifications (typically 1-2 hours at 37°C).
- Purify the digested products to remove enzymes and buffers.
Step 3: Ligate Insert into Vector
- Mix the digested insert and vector in a molar ratio (e.g., 3:1 insert:vector) with ATP-dependent DNA ligase (e.g., T4 DNA Ligase) and an appropriate buffer.
- Incubate the ligation reaction at room temperature for 1 hour or 16°C overnight.
Step 4: Transform into Bacterial Cells
- Introduce the ligation product into competent E. coli cells via heat shock or electroporation.
- Plate the transformed bacteria onto agar plates containing a selective antibiotic (e.g., ampicillin).
Step 5: Screen for Recombinant Clones
- After overnight growth, select individual colonies for screening.
- Use colony-PCR with insert-specific primers to identify colonies containing the recombinant plasmid.
- Culture positive clones and purify the plasmid DNA for sequence verification.
Application Notes and In Vivo Protocol
The true power of Chronos and Chrimson is realized in their application to probe neural circuits in living brain tissue. The following protocol describes a method for validating opsin function and achieving two-color stimulation in acute brain slices, a critical step before in vivo experiments.
Protocol: Two-Color Neural Stimulation in Acute Brain Slice
Objective: To independently stimulate two distinct neural populations expressing Chronos and Chrimson in a single acute brain slice preparation, while recording postsynaptic responses.
Materials and Reagents:
- Acute brain slices from transgenic or virally-injected mice.
- Artificial Cerebrospinal Fluid (aCSF).
- Laser systems or LEDs for 470 nm (blue) and 590 nm (red) light delivery.
- Patch-clamp electrophysiology setup.
Procedure:
- Animal Preparation and Viral Injection:
- Anesthetize the mouse and secure it in a stereotaxic frame.
- Perform a craniotomy at the coordinates of the target brain region (e.g., primary motor cortex).
- Use a microinjection system (e.g., Hamilton syringe) to deliver AAVs containing Chronos under a cell-type-specific promoter (e.g., CaMKIIα for excitatory neurons) into one region, and AAVs containing Chrimson under a different promoter (e.g., hSyn for a broader population) into an adjacent or contralateral region [46]. Allow 3-6 weeks for opsin expression.
Acute Slice Preparation:
- Anesthetize and transcardially perfuse the mouse with ice-cold, sucrose-based aCSF saturated with 95% O₂ / 5% CO₂.
- Decapitate, rapidly extract the brain, and prepare 300 μm thick coronal slices using a vibratome.
- Recover slices in standard aCSF at 34°C for 30 minutes, then maintain at room temperature.
Dual-Optogenetic Stimulation and Recording:
- Transfer a single slice to the recording chamber, continuously perfused with oxygenated aCSF.
- Patch a postsynaptic neuron in a downstream target region (e.g., striatum).
- To stimulate Chronos-expressing neurons, deliver 5 ms pulses of blue light (470 nm) at a low irradiance (≤ 1 mW/mm²). This irradiance should be subthreshold for Chrimson activation [46].
- To stimulate Chrimson-expressing neurons, deliver 10-15 ms pulses of red light (590 nm).
- Interleave blue and red light stimuli to demonstrate independent activation of the two neural pathways without crosstalk.
Troubleshooting:
- Crosstalk (Blue light activates Chrimson neurons): Reduce the blue light irradiance. The high red-shift of Chrimson naturally minimizes its blue-light sensitivity, but high power can overcome this [46].
- Poor Spike Fidelity at High Frequency: For Chronos, ensure kinetic properties are fast enough. For Chrimson, use the ChrimsonR variant for frequencies above 10 Hz [46].
The experimental workflow for viral delivery and validation is summarized below.
The Scientist's Toolkit: Research Reagent Solutions
Successful optogenetics experiments require a suite of carefully selected reagents. The table below catalogs essential materials and their functions.
Table 2: Essential Research Reagents for Optogenetic Experiments
| Reagent / Material | Function | Example Tools & Notes |
|---|---|---|
| Opsin Plasmid DNA | Core light-sensitive actuator. | Chronos (fast, blue-green), Chrimson/ChrimsonR (red-shifted); available from Addgene [47]. |
| Viral Vector | Efficient in vivo gene delivery. | Adeno-Associated Virus (AAV, e.g., serotype 2/8 or 2/9 for neurons); Lentivirus. |
| Cell-Type-Specific Promoter | Targets opsin expression to defined neural populations. | CaMKIIα (excitatory neurons), GAD67 (inhibitory neurons), hSyn (pan-neuronal). |
| Fluorescent Reporter | Visualizes transfected/infected cells. | Fused in-frame with the opsin (e.g., EYFP, mCherry) or expressed from an IRES/T2A sequence. |
| Restriction Enzymes / Cloning Kit | Molecular construction of plasmid vectors. | EcoRI, NotI; Gibson Assembly Master Mix; Gateway BP/LR Clonase. |
| Competent Bacterial Cells | Plasmid amplification. | High-efficiency E. coli strains (e.g., DH5α, Stbl3 for AAV plasmids). |
The application of rDNA technology in optogenetics continues to evolve, pushing the boundaries of neuroscience research. Current efforts focus on engineering next-generation opsins with enhanced properties, such as HulaChrimson, a recently discovered variant with high sequence similarity to Chrimson but a significantly blue-shifted action spectrum, providing new insights into the molecular mechanisms of color-tuning [52]. Furthermore, the translation of optogenetic tools into therapeutic strategies is underway, with clinical trials exploring optogenetic vision restoration for retinal degenerative diseases [53]. The NIH BRAIN Initiative actively funds the development and validation of novel tools for cell-specific and circuit-specific processes, underscoring the critical role of continued rDNA innovation in neuroscience [54].
In conclusion, the synergy between recombinant DNA technology and optogenetics has provided neuroscientists with an unparalleled set of tools for deconstructing the brain's complex wiring. Chronos and Chrimson represent a significant leap forward, enabling precise, multi-color control over neural activity. The protocols and resources detailed in this Application Note provide a roadmap for researchers to effectively employ these tools, driving discovery in basic neuroscience and paving the way for novel therapeutic interventions.
Molecular cloning and recombinant DNA technology serve as foundational tools in modern neuroscience research, enabling the precise dissection of disease mechanisms and the development of innovative therapeutic strategies. By allowing scientists to isolate, amplify, and manipulate specific genes, these techniques provide a direct window into the molecular machinery of neurodegeneration, oncogenesis, and synaptic function. This application note details how these technologies drive progress in understanding and treating complex neurological conditions, featuring established protocols and key reagent solutions to accelerate research in Alzheimer's disease, glioblastoma, and synaptic plasticity.
Application Note 1: Plasmalogen Synthesis Gene TMEM189 in Alzheimer's Disease
Background and Rationale
Plasmalogens, a unique class of ether phospholipids, play a crucial role in neuronal membrane integrity, antioxidant defense, and cellular signaling within the brain. A significant body of evidence now links abnormal plasmalogen metabolism to the pathogenesis of Alzheimer's disease (AD), with levels of these protective lipids declining with age and during disease progression [55] [56]. The gene tmem189, which encodes the enzyme plasmanylethanolamine desaturase, has been identified as a critical final step in plasmalogen biosynthesis. Molecular cloning of this gene enables functional studies to elucidate its role in AD pathology and explore its potential as a therapeutic target [55].
Key Experimental Findings
Recent research utilizing molecular cloning techniques has yielded critical insights into TMEM189 function:
- Molecular Characterization: The open reading frame (ORF) of the tmem189 cDNA from Takifugu rubripes was cloned and sequenced, revealing a 828 bp sequence encoding a 275-amino acid protein. Bioinformatics analysis predicted four transmembrane domains, suggesting its function as a transmembrane protein [55] [56].
- Tissue-Specific Expression: Analysis in T. rubripes demonstrated higher tmem189 expression in brain tissue compared to other tissues, indicating its particular importance in the nervous system [55].
- Functional Validation via Overexpression: Researchers transfected a GFP-tagged tmem189 eukaryotic expression vector into HEK293T cells, confirming successful protein expression. Subsequent liquid chromatography-mass spectrometry (LC-MS) analysis demonstrated that tmem189 overexpression directly promotes plasmalogen synthesis, establishing a direct functional link between the cloned gene and lipid production [55].
Table 1: Key Quantitative Data from TMEM189 Cloning and Functional Analysis
| Parameter | Finding | Significance |
|---|---|---|
| ORF Length | 828 bp | Confirms gene sequence for further manipulation [56] |
| Amino Acids | 275 | Determines basic protein structure [56] |
| Molecular Mass | 31.41 kDa | Informs protein analysis experiments [56] |
| Isoelectric Point | 6.67 | Critical for protein purification strategies [56] |
| Key Functional Result | Increased plasmalogen synthesis in transfected cells | Validates TMEM189's role in plasmalogen pathway [55] |
Detailed Protocol: Cloning and Functional Analysis of TMEM189
Objective: To clone the TMEM189 gene, express it in a mammalian cell line, and functionally validate its role in plasmalogen synthesis.
Materials & Reagents:
- cDNA library from target tissue (e.g., T. rubripes brain)
- PCR reagents (polymerase, dNTPs, buffers)
- Specific primers for TMEM189 ORF
- Eukaryotic expression vector (e.g., pEGFP-N1 for GFP-tagging)
- Restriction enzymes and T4 DNA Ligase
- HEK293T cells
- Cell culture media and transfection reagent
- Liquid Chromatography-Mass Spectrometry (LC-MS) system
Procedure:
Gene Amplification:
- Design forward and reverse primers with overhangs containing appropriate restriction sites.
- Amplify the full-length TMEM189 ORF via PCR using high-fidelity DNA polymerase.
- Purify the PCR product and digest both the insert and the chosen expression vector with the selected restriction enzymes.
Ligation and Transformation:
- Ligate the digested TMEM189 fragment into the prepared vector using T4 DNA Ligase.
- Transform the ligation product into competent E. coli cells and plate on selective antibiotic media.
- Select colonies, culture them, and isolate plasmid DNA for verification by restriction digest and Sanger sequencing.
Cell Transfection and Expression:
- Culture HEK293T cells under standard conditions (DMEM, 10% FBS, 37°C, 5% CO2).
- Transfect the verified TMEM189 plasmid into the cells using a suitable transfection reagent (e.g., polyethyleneimine or calcium phosphate).
- Confirm transfection efficiency and protein localization via fluorescence microscopy (if using a GFP-tagged construct).
Functional Validation - Plasmalogen Measurement:
- Harvest transfected cells 48-72 hours post-transfection.
- Extract lipids from the cell pellet using a methanol-chloroform protocol.
- Analyze plasmalogen species using LC-MS. Compare the lipid profiles of TMEM189-transfected cells to empty vector-transfected controls.
- Quantify specific plasmalogen peaks (e.g., plasmenylethanolamine) to confirm the functional role of TMEM189.
Signaling Pathway and Workflow Diagram
Diagram 1: Experimental workflow for cloning and validating TMEM189 function.
Application Note 2: Molecular Targeting in Glioblastoma
Background and Rationale
Glioblastoma (GBM) is the most aggressive and lethal primary brain tumor in adults, characterized by profound molecular heterogeneity, therapeutic resistance, and a dismal median survival of 12-15 months [57]. Molecular cloning technologies are instrumental in classifying GBM subtypes based on genetic drivers and developing targeted therapies. Key oncogenic alterations include amplifications in the epidermal growth factor receptor (EGFR), platelet-derived growth factor receptor (PDGFR), and dysregulation of the PI3K/AKT/mTOR pathway [57]. Recent research has moved beyond traditional kinase targets to explore novel vulnerabilities, such as the role of non-muscle myosin motors in tumor invasion and treatment resistance [58].
Key Experimental Findings and Therapeutic Intervention
- Molecular Classification: GBM is subtyped based on transcriptional profiles: Proneural, Neural, Classical, and Mesenchymal, each with distinct genetic features and clinical outcomes. The Mesenchymal subtype, associated with the worst prognosis, is characterized by NF1 and PTEN loss and activation of pro-inflammatory and angiogenic pathways [57].
- Myosin Motor Targeting: The experimental drug MT-125, a first-in-class myosin motor inhibitor, demonstrates a multi-faceted anti-tumor effect in GBM models. It sensitizes resistant cancer cells to radiation, induces multinucleation and cell death, and blocks cellular invasion [58].
- Synergistic Therapy: Combining MT-125 with existing chemotherapy drugs like sunitinib (a kinase inhibitor) produces a powerful, sustained disease-free state in mouse models of glioblastoma, an effect not previously observed with single-agent therapies [58].
Table 2: Key Findings from Novel Glioblastoma Therapeutic Strategy (MT-125)
| Therapeutic Effect | Mechanism | Experimental Evidence |
|---|---|---|
| Re-sensitizes to Radiation | Renders malignant cells newly sensitive to radiation therapy. | Animal studies showing enhanced radiation response [58] |
| Inhibits Cell Division | Generates multinucleated cells that cannot complete division. | Cells marked for death due to failed cytokinesis [58] |
| Blocks Tumor Invasion | Inhibits cell motility by targeting myosin motors. | Prevents cells from squeezing and changing shape [58] |
| Synergy with Chemotherapy | Potentiates effects of kinase inhibitors. | Long disease-free states in mice when combined with sunitinib [58] |
Detailed Protocol: Evaluating Myosin Inhibitor Efficacy in GBM Models
Objective: To test the efficacy of myosin inhibitor MT-125, alone and in combination with standard therapies, in glioblastoma models.
Materials & Reagents:
- Patient-derived glioblastoma stem-like cells (GSCs) or established GBM cell lines.
- Myosin inhibitor (e.g., MT-125)
- Chemotherapy drugs (e.g., sunitinib)
- Cell culture materials and invasion assay kits (e.g., Matrigel-coated Transwells)
- Irradiator for in vitro radiation studies
- Apoptosis detection kit (Annexin V/PI)
- Immunofluorescence staining reagents (DAPI, phalloidin)
Procedure:
In Vitro Combination Treatment:
- Culture GBM cells in appropriate media. Seed cells in 96-well plates.
- Treat cells with a dose matrix of MT-125 and a chemotherapeutic agent (e.g., sunitinib) for 72 hours.
- Assess cell viability using a colorimetric assay (e.g., MTT or CellTiter-Glo). Calculate the combination index (CI) using software like CompuSyn to determine synergy (CI<1), additivity (CI=1), or antagonism (CI>1).
Radiation Sensitivity Assay:
- Pre-treat GBM cells with a sub-lethal dose of MT-125 for 24 hours.
- Expose cells to varying doses of radiation (e.g., 2, 4, 6 Gy).
- Conduct clonogenic survival assays: allow cells to grow for 10-14 days, then fix, stain with crystal violet, and count colonies. Plot survival fractions to determine if MT-125 enhances radiation sensitivity.
Invasion Assay:
- Pre-treat cells with MT-125 for 24 hours.
- Seed the treated cells into the upper chamber of a Matrigel-coated Transwell insert in serum-free medium. Place complete medium with serum in the lower chamber as a chemoattractant.
- After 24-48 hours, fix the cells, stain them, and wipe the non-invaded cells from the top membrane. Count the invaded cells on the bottom membrane under a microscope. Compare to untreated controls.
Mode of Cell Death Analysis:
- Treat GBM cells with MT-125 and/or other drugs for 24-48 hours.
- Harvest cells and stain with Annexin V-FITC and Propidium Iodide (PI).
- Analyze by flow cytometry to distinguish live (Annexin V-/PI-), early apoptotic (Annexin V+/PI-), late apoptotic (Annexin V+/PI+), and necrotic (Annexin V-/PI+) populations.
Signaling Pathway and Experimental Flow Diagram
Diagram 2: Mechanism of action for myosin inhibitor MT-125 in glioblastoma.
Application Note 3: Probing Synaptic Plasticity in Neurodegeneration
Background and Rationale
Synaptic plasticity—the activity-dependent change in the strength of communication between neurons—is the fundamental cellular mechanism underlying learning, memory, and behavioral adaptation. Its dysfunction is a core pathological feature of both Alzheimer's disease and other neuropsychiatric disorders [59]. Advanced tools, including genetically encoded voltage indicators (GEVIs) and computational models, now allow for unprecedented quantification of synaptic dynamics in health and disease. Furthermore, investigations into how neuromodulators, including psychedelics, influence plasticity rules are opening new therapeutic avenues [60] [59].
Key Experimental Findings
- All-Optical Interrogation of Synapses: A novel all-optical approach using JEDI-2Psub, a GEVI optimized for detecting subthreshold voltage changes, enables the measurement of synaptic potentials and long-term plasticity at identified cerebellar synapses in awake, behaving mice. This allows for the correlation of voltage signals within and between neighboring neurons during behavior [60].
- Reversal of Plasticity Polarity by Psychedelics: In the claustrum—a brain region with high density of serotonin 2A receptors (5-HT2AR)—the psychedelic drug DOI was found to reverse the polarity of synaptic plasticity. Under control conditions, a standard pre-post stimulation protocol induced long-term depression (LTD). However, in the presence of DOI, the same protocol induced long-term potentiation (LTP), demonstrating a drug-induced metaplasticity effect [59].
- Computational Modeling of Plasticity: The SRPlasticity software package provides a flexible computational framework for characterizing short-term plasticity (STP) from electrophysiological data. It uses a Spike Response Plasticity (SRP) model to infer key parameters of synaptic dynamics and predict responses to novel stimulus patterns, helping to classify functional synaptic types [61].
Detailed Protocol: All-Optical Probing of Synaptic Plasticity In Vivo
Objective: To measure and manipulate synaptic plasticity at identified synapses in the brain of an awake, behaving mouse using a combination of voltage imaging, optogenetics, and sensory stimulation.
Materials & Reagents:
- Mice (e.g., Math1-Cre for cerebellar granule cell targeting)
- AAVs: CaMKII-JEDI-2Psub (for Purkinje cell expression) and Cre-dependent ChRmine-mScarlet (for granule cell expression)
- Stereotaxic surgery equipment
- Two-photon microscope with resonant scanners and integrated laser for optogenetics (e.g., 635 nm LED)
- Head-plate for animal immobilization
- Sensory stimulation setup (e.g., air-puff for whisker pad)
Procedure:
Viral Injection and Surgery:
- Anesthetize the mouse and secure it in a stereotaxic frame.
- Co-inject AAV-CaMKII-JEDI-2Psub and AAV-FLEX-ChRmine-mScarlet into the cerebellar vermis (Lobules V/VI) of Math1-Cre mice. This ensures GEVI expression in Purkinje cells (PCs) and opsin expression in granule cells (GrCs).
- Implant a cranial window above the injection site and attach a head-plate for future head-fixing. Allow 3-4 weeks for viral expression.
Two-Photon Voltage Imaging and Optogenetics:
- Head-fix the awake mouse on a running wheel under the two-photon microscope.
- Image JEDI-2Psub fluorescence in PC dendrites at a high frame rate (e.g., 440 Hz) using a wavelength of ~920 nm.
- Record baseline dendritic voltage activity, capturing spontaneous complex spikes.
Sensory and Optogenetic Stimulation:
- Deliver brief air-puffs to the whisker pad to evoke sensory-driven climbing fiber (CF) responses in PCs. Record the resulting voltage transients.
- Selectively activate GrC inputs by delivering light pulses (e.g., 5-20 ms) through the microscope objective to activate ChRmine. This evokes parallel fiber inputs to PCs, typically resulting in measurable inhibitory postsynaptic potentials (IPSPs).
Plasticity Induction and Measurement:
- To induce plasticity, pair the optogenetic activation of GrCs (presynaptic stimulus) with the sensory-evoked CF input (postsynaptic stimulus) with a specific timing.
- Repeatedly apply this pairing protocol over several minutes.
- Continuously monitor the amplitude of the optogenetically-evoked IPSPs in the PC dendrites before and after the pairing protocol to quantify long-term changes in synaptic strength (e.g., LTP or LTD of inhibition).
Signaling Pathway and Experimental Flow Diagram
Diagram 3: All-optical workflow for probing synaptic plasticity in vivo.
The Scientist's Toolkit: Essential Research Reagents & Tools
Table 3: Key Research Reagent Solutions for Molecular Cloning and Neuroscience Applications
| Reagent/Tool | Core Function | Example Application |
|---|---|---|
| Eukaryotic Expression Vectors | Plasmid backbone for gene delivery and protein expression in mammalian cells. | Cloning TMEM189 for overexpression and functional study in HEK293T cells [55]. |
| Genetically Encoded Voltage Indicators (GEVIs) | Fluorescent protein-based sensors for reporting changes in membrane potential. | Monitoring postsynaptic potentials in Purkinje cell dendrites in vivo (e.g., JEDI-2Psub) [60]. |
| Channelrhodopsins & Optogenetic Tools | Light-activated ion channels for precise temporal control of neuronal activity. | Selective activation of granule cell inputs to probe cerebellar circuitry (e.g., ChRmine) [60]. |
| AAV Delivery Vectors | Adeno-associated viruses for efficient and specific gene delivery in the brain. | Cell-type-specific expression of GEVIs and opsins in defined neuronal populations [60]. |
| SRPlasticity Software | Computational model for characterizing and predicting short-term synaptic plasticity. | Automated fitting of electrophysiology data to infer synaptic parameters and classify STP [61]. |
| Myosin Motor Inhibitors | First-in-class small molecules targeting non-muscle myosin II. | Disrupting glioblastoma cell division, motility, and therapy resistance (e.g., MT-125) [58]. |
Channelrhodopsins (ChRs), light-gated ion channels derived from microbial opsins, have revolutionized neuroscience by enabling precise optical control of neuronal activity. This application note details the molecular engineering of recombinant Channelrhodopsins, framing these techniques within the broader context of molecular cloning and recombinant DNA technology. As optogenetics expands into complex applications such as multiplexed neuronal control and clinical therapeutics, the demand for ChRs with enhanced properties—including improved spectral characteristics, ion selectivity, and subcellular targeting—has intensified [62] [63]. Traditional approaches of simply expressing wild-type ChRs are insufficient for advanced applications, necessitating sophisticated protein engineering strategies.
This document provides a comprehensive technical resource for researchers and drug development professionals, detailing the key principles, methods, and validation protocols for engineering next-generation Channelrhodopsins. We place particular emphasis on strategies for achieving spectral separation for multiplexed optogenetics, optimizing membrane trafficking to maximize photocurrents, and implementing subcellular targeting for circuit-specific interrogation [64] [65]. The protocols outlined herein have direct applications in both basic research, for deconstructing complex neural circuits, and in translational medicine, particularly for sensory restoration therapies in blindness and deafness [64] [66] [63].
Key Engineering Strategies and Quantitative Comparison
The engineering of recombinant Channelrhodopsins focuses on modifying core biophysical and cellular properties to suit specific experimental or therapeutic needs. Table 1 summarizes the performance characteristics of several key natural and engineered ChR variants, highlighting the trade-offs between spectral properties, conductance, and kinetic properties that must be considered during the design phase.
Table 1: Performance Characteristics of Selected Natural and Engineered Channelrhodopsins
| Channelrhodopsin Variant | Spectral Peak (nm) | Ion Selectivity | Relative Photocurrent Amplitude | Key Engineering Feature | Primary Application |
|---|---|---|---|---|---|
| AnsACR (Natural) | ~440 [62] | Cl⁻ [62] | High (Robust) [62] | Natural blue-shift [62] | Neuronal silencing, multiplexed optogenetics [62] |
| NlCCR (Natural) | ~440 [62] | Cations (H⁺, Na⁺) [62] | High (Exceeds prior tools) [62] | Natural blue-shifted CCR [62] | Neuronal activation with blue light [62] |
| f-Chrimson | ~590 [64] | Cations [64] | Baseline (with EYFP tag) [64] | Red-shifted, fast kinetics [64] | Deep-tissue activation, auditory nerve stimulation [64] |
| f-Chrimson-TSKir2.1 | ~590 [64] | Cations [64] | ~82% of f-Chrimson-EYFP (25.5 → 21.0 pA/pF) [64] | Trafficking signal replacement [64] | Clinical translation (enhanced trafficking) [64] |
| ChR2 XXM2.0 | ~460 [67] | Ca²⁺ (High) [67] | Highest among Ca²⁺-permeable ChRs [67] | Enhanced Ca²⁺ conductance [67] | Subcellular Ca²⁺ signaling manipulation [67] |
A critical consideration in clinical translation is minimizing immunogenicity and potential cellular dysfunction. A 2024 study demonstrated that overexpression of ChR2-EYFP in skeletal muscle caused significant contractile dysfunction and downregulation of genes related to transmembrane transport, while "ChR2-only" constructs (lacking the fluorescent protein) did not [68]. This finding underscores the importance of tag-free or alternative-tagging strategies for therapeutic applications.
Detailed Experimental Protocols
Protocol 1: Trafficking Optimization via C-Terminal Tag Engineering
Background: A major hurdle in clinical translation is the reduction of photocurrent amplitude upon removal of the C-terminal fluorescent protein (FP) tag, which is desirable to avoid potential immunogenicity and cellular toxicity [64] [68]. This protocol describes the replacement of the FP with a specialized trafficking sequence to restore plasma membrane expression and function.
Methodology:
Vector Construction:
- Using standard molecular cloning techniques (e.g., restriction enzyme digestion/ligation or Gibson assembly), create the following constructs for comparative analysis:
- Control: f-Chrimson-EYFP (full-length) [64].
- Test Group 1: f-Chrimson* (EYFP removed) [64].
- Test Group 2: f-Chrimson-TSKir2.1-P2A-Katushka. Precisely fuse the 20-amino acid plasma membrane trafficking sequence of the human potassium channel Kir2.1 (TSKir2.1) to the C-terminus of the tag-free f-Chrimson. Incorporate a P2A self-cleaving peptide sequence followed by the red fluorescent protein Katushka to enable transfection visualization and selection [64].
- Clone all final constructs into an appropriate adeno-associated virus (AAV) vector backbone under a strong, ubiquitous promoter (e.g., CAG or CBI) for subsequent functional testing.
- Using standard molecular cloning techniques (e.g., restriction enzyme digestion/ligation or Gibson assembly), create the following constructs for comparative analysis:
In Vitro Functional Validation:
- Transiently transfect the constructed plasmids into a mammalian cell line such as NG cells or HEK293T cells.
- After 48-72 hours, perform whole-cell patch-clamp electrophysiology.
- Hold cells at a physiological membrane potential (e.g., -60 mV) and illuminate with a 593 nm LED/laser at saturating intensity (e.g., 21 mW/mm²) for a 500 ms pulse.
- Record the peak photocurrent density (pA/pF) by normalizing the current amplitude to cell capacitance.
- Expected Outcome: The f-Chrimson-TSKir2.1 construct should recover ~80% of the photocurrent density of the FP-tagged control, a significant improvement over the tag-free f-Chrimson*, which typically shows a >75% reduction [64].
In Vivo Application:
- Package the f-Chrimson-TSKir2.1 construct into AAV2/9 particles for in vivo use.
- Stereotaxically inject the virus into the target tissue (e.g., mouse spiral ganglion for auditory research) [64].
- After 3-6 weeks for sufficient expression, validate functional rescue using techniques such as recordings of optically evoked auditory brainstem responses (oABRs) [64].
Diagram 1: Workflow for trafficking optimization.
Protocol 2: Engineering Axon-Terminal Specific Channelrhodopsins
Background: Precise neural circuit mapping requires optogenetic tools that can selectively stimulate synaptic outputs without activating passing fibers or somatodendritic compartments. This protocol utilizes a subcellular targeting tag to direct ChR2 expression specifically to axon terminals [65].
Methodology:
Tag Selection and Fusion:
- Fuse the C-terminal domain (amino acids 820-872) of the metabotropic glutamate receptor 2 (mGluR2), which naturally localizes to presynaptic terminals, to the C-terminus of ChR2(H134R)-EYFP.
- To further enhance specificity, downstream of the mGluR2 sequence, add a PEST proteolytic motif (promotes degradation in somatodendritic compartments) and an axon-targeting element (ATE). This combined tag is termed "mGluR2-PA" [65].
Validation in Cultured Neurons:
- Express the constructed ChR2-YFP-mGluR2-PA in primary hippocampal neurons via AAV-DJ transduction.
- After 14-21 days, fix the cultures and immunostain for markers of the axon (Tau), dendrites (MAP2), and presynaptic active zones (Bassoon).
- Perform high-resolution confocal microscopy and quantify colocalization using Pearson's correlation coefficient.
- Expected Outcome: The mGluR2-PA tag should significantly increase colocalization with Bassoon (presynaptic) and Tau (axonal), while drastically reducing colocalization with MAP2 (dendritic) compared to untagged ChR2-YFP [65].
Functional In Vivo Circuit Mapping:
- Inject AAV vectors carrying ChR2-YFP-mGluR2-PA into a specific brain region (e.g., hippocampal CA3).
- In the contralateral projection area, use a recording electrode to detect antidromic spikes evoked by light stimulation of the axon terminals.
- Apply the Spike Collision Test to confirm the antidromic nature of the evoked spikes, which validates the presynaptic-specific activation and reduces polysynaptic contamination [65].
Diagram 2: Presynaptic targeting strategy.
The Scientist's Toolkit: Research Reagent Solutions
Successful engineering and application of recombinant Channelrhodopsins rely on a suite of specialized reagents. Table 2 catalogues the essential materials and their functions for the protocols described in this document.
Table 2: Essential Research Reagents for Channelrhodopsin Engineering
| Reagent / Material | Function / Purpose | Example Use Case |
|---|---|---|
| AAV Vectors (e.g., Serotype 2/9, DJ) | Efficient gene delivery vehicle for in vitro and in vivo expression of ChR constructs. Shows high neuronal tropism and relatively low immunogenicity [64] [66]. | Widespread use in neuronal transduction for basic research and clinical trials [64] [63]. |
| TSKir2.1 Trafficking Sequence | A 20-aa peptide from the inward rectifying K+ channel Kir2.1 that enhances forward trafficking of fused membrane proteins to the plasma membrane [64]. | Replaces fluorescent protein tags (e.g., EYFP) to boost photocurrents in clinical candidate opsins like f-Chrimson [64]. |
| mGluR2-PA Localization Tag | A composite tag (mGluR2 C-terminal + PEST + Axon Targeting Element) that directs protein expression specifically to presynaptic axon terminals [65]. | Creating circuit-specific optogenetic tools for mapping long-range axonal projections without somatic contamination [65]. |
| Ancyromonad Channelrhodopsins (e.g., AnsACR, NlCCR) | A class of naturally occurring, potent blue-shifted (~440 nm) ChRs with either anion or cation selectivity [62] [69]. | Provides spectrally separated, high-amplitude actuators for multiplexed all-optical electrophysiology [62]. |
| ChR2 XXM2.0 | An engineered ChR2 variant with a point mutation (H134Q) and signal peptides, conferring high Ca²⁺ conductance and membrane expression [67]. | Optogenetic induction of subcellular Ca²⁺ signals in megakaryocytes and platelets to study calcium-dependent processes [67]. |
| P2A Self-Cleaving Peptide | A short peptide sequence that induces "ribosomal skipping," allowing co-expression of multiple proteins (e.g., opsin and a fluorescent reporter) from a single mRNA transcript [64]. | Enables visualization of successfully transduced cells (via Katushka) while expressing a tag-free opsin, minimizing fusion protein artifacts [64]. |
The engineering of recombinant Channelrhodopsins represents a sophisticated application of molecular cloning and recombinant DNA technology, directly addressing the evolving needs of modern neuroscience. As demonstrated, strategic modifications—such as incorporating trafficking signals, subcellular targeting tags, and leveraging natural diversity—can profoundly enhance the specificity, efficiency, and safety of optogenetic tools. The protocols and data summarized here provide a foundational roadmap for researchers developing novel actuators for dissecting neural circuits or creating next-generation gene therapies for neurological and sensory disorders. The continued synergy between protein engineering, viral vectorology, and electrophysiology will undoubtedly yield even more powerful and precise tools for the control and understanding of brain function.
Navigating the Lab Bench: Troubleshooting Common Cloning Challenges in Neuroscience Experiments
Molecular cloning is a foundational technique in modern bioscience research, enabling the isolation, replication, and expression of specific DNA sequences. This process of creating recombinant DNA has revolutionized biological research and medicine, forming the cornerstone of countless applications from basic gene function studies to the development of novel therapeutics [22] [70]. For neuroscience research, molecular cloning provides indispensable tools for investigating neural gene function, expressing neuronal proteins, and developing models of neurological disease.
The core principle of molecular cloning involves introducing a DNA fragment of interest into a self-replicating genetic vector, which is then propagated in a host organism, most commonly the bacterium E. coli [71]. This allows researchers to amplify specific neural genes or regulatory elements for detailed study. The resulting recombinant DNA molecules can be used to express proteins for structural analysis, create cellular models of neurological disorders, or engineer viral vectors for gene therapy approaches to treat neurodegenerative conditions.
This application note details a standardized five-step cloning workflow, providing neuroscience researchers with a comprehensive roadmap from initial fragment generation to final analysis of cloned constructs, with particular emphasis on protocols and reagents optimized for neural gene studies.
The Five-Step Workflow: A Detailed Roadmap
The molecular cloning process can be systematically divided into five critical stages, each requiring specific reagents and quality control checkpoints. The following workflow diagram illustrates the complete process and key decision points.
Step 1: Fragment Generation
The initial step involves preparing the DNA fragment of interest (insert) and the cloning vector for subsequent ligation. For neuroscience applications, inserts may include genes encoding neuronal receptors, ion channels, synaptic proteins, or regulatory elements controlling neural-specific expression.
Insert Preparation Methods
- Restriction Enzyme Digestion: Uses sequence-specific endonucleases to create compatible ends on both insert and vector [72] [73]. For neuronal genes, careful selection of enzymes that don't cut within critical functional domains is essential.
- PCR Amplification: Amplifies target sequences from genomic DNA, cDNA, or existing plasmids [71]. Reverse transcriptase PCR (RT-PCR) is particularly valuable for neuroscience to create cDNA copies of neuronal mRNAs.
- Gene Synthesis: Artificial synthesis of DNA sequences based on desired nucleotide sequence [22]. This approach allows for codon optimization for enhanced expression of human neuronal proteins in model systems.
Vector Preparation
The cloning vector must be prepared to receive the insert through restriction enzyme digestion. Dephosphorylation of the vector ends using alkaline phosphatase is often performed to prevent self-ligation [72]. Vectors containing neural-specific promoters (e.g., synapsin, CaMKIIα) are particularly valuable for neuroscience applications.
Table 1: Common Restriction Enzymes for Fragment Generation
| Enzyme | Recognition Sequence | Overhang Type | Common Applications in Neuroscience |
|---|---|---|---|
| EcoRI | 5'-G↓AATTC-3' | 5' overhang | General cDNA cloning |
| BamHI | 5'-G↓GATCC-3' | 5' overhang | Insertion into neural expression vectors |
| NotI | 5'-GC↓GGCCGC-3' | 5' overhang | Cloning large genomic fragments for YAC libraries |
| KpnI | 5'-GGTAC↓C-3' | 3' overhang | Directional cloning of promoter elements |
Step 2: Cloning
This critical step involves joining the prepared insert and vector fragments to create a recombinant DNA molecule. The choice of cloning method depends on the specific requirements of the neuroscience research project.
Restriction Enzyme-Based Cloning
The traditional method uses restriction enzymes and DNA ligase to join complementary ends [72] [70]. The enzyme T4 DNA ligase catalyzes the formation of phosphodiester bonds between adjacent 5'-phosphate and 3'-hydroxyl groups of DNA fragments [72]. A typical reaction setup includes:
Protocol: Mix components gently and incubate at 14-25°C for 10 minutes to 16 hours, depending on required yield [72]. For blunt-ended ligations, add 5% PEG to improve efficiency. Heat inactivation is not recommended when using PEG.
Advanced Cloning Techniques
- Gateway Recombination: Uses site-specific recombination rather than restriction enzymes, allowing rapid transfer of DNA sequences between different vectors [74]. Ideal for creating multiple neural expression constructs from a single validated cDNA.
- Gibson Assembly: Isothermal assembly method that uses 5' exonuclease, polymerase, and ligase to join multiple fragments with homologous ends [74]. Enables assembly of large neuronal gene circuits.
- TA Cloning: Leverages the terminal transferase activity of Taq polymerase that adds a single 3'A overhang to PCR products, compatible with T-overhang vectors [74].
Step 3: Transformation
Transformation introduces the recombinant DNA molecules into competent bacterial cells (usually E. coli) for propagation [72] [70]. The transformation efficiency is a critical factor in cloning success, especially for complex libraries or large constructs.
Transformation Protocols
Chemical Transformation Method:
- Thaw chemically competent E. coli cells on ice.
- Add 1-5 μL ligation reaction to 50-100 μL competent cells, mix gently.
- Incubate on ice for 20-30 minutes.
- Heat shock at 42°C for 30-45 seconds without shaking.
- Immediately return to ice for 2 minutes.
- Add 250-500 μL recovery medium.
- Incubate at 37°C with shaking for 45-60 minutes.
- Plate onto selective agar plates containing appropriate antibiotic.
Electroporation Method:
- Use electrocompetent cells prepared with multiple washes in cold water or buffer.
- Mix DNA with cells in cold electroporation cuvette.
- Apply electrical pulse (typically 1.8-2.5 kV for E. coli).
- Immediately add recovery medium.
- Incubate with shaking for 45-60 minutes before plating.
Table 2: Competent Cell Selection Guide
| Cell Strain | Transformation Efficiency | Key Features | Neuroscience Applications |
|---|---|---|---|
| DH5α | 1 x 10⁷ - 10⁸ CFU/μg | General cloning, blue-white screening | Routine plasmid propagation |
| TOP10 | 1 x 10⁹ CFU/μg | High efficiency, recA1 mutation | Library construction, difficult clones |
| BL21(DE3) | 1 x 10⁸ CFU/μg | Protein expression, T7 RNA polymerase | Neuronal protein expression |
| Stbl3 | 5 x 10⁷ CFU/μg | Stabilizes repetitive sequences | Lentiviral production for neuronal transduction |
| JM107 | 1 x 10⁸ CFU/μg | lacZΔM15 for blue-white screening | cDNA library screening |
Step 4: Selection and Screening
After transformation, selective pressure is applied to identify cells containing recombinant plasmids, followed by screening to identify correct clones [70].
Selection Methods
Antibiotic resistance genes in the vector allow only bacteria containing plasmids to grow [70] [71]. Common selection antibiotics include ampicillin (50-100 μg/mL), kanamycin (25-50 μg/mL), and chloramphenicol (25-170 μg/mL).
Screening Techniques
- Blue-White Screening: Uses the lacZα gene complementation system [72] [70]. Successful insertion of DNA into the multiple cloning site disrupts the lacZα gene, resulting in white colonies rather than blue.
- Colony PCR: Screens colonies directly by PCR using insert-specific or vector-specific primers [71].
- Restriction Analysis: Small-scale plasmid preparations (minipreps) followed by restriction enzyme digestion to verify insert size [70].
Step 5: Analysis
Final verification ensures the cloned construct matches the expected sequence and structure before use in neuroscience experiments.
Analytical Methods
- Restriction Digest Analysis: Confirms insert presence and orientation using specific restriction enzymes [70].
- Sequencing: Provides complete sequence verification [70]. Sanger sequencing is standard; next-generation sequencing may be used for complex constructs.
- Analytical PCR: Amplifies the insert region to verify size and identity.
Quantitative Analysis
For neuroscience applications requiring precise expression levels, additional analyses may include:
- Copy number determination using quantitative PCR
- Transcript expression analysis by Northern blot or RT-PCR
- Protein expression verification by Western blot when using expression vectors
The Scientist's Toolkit: Essential Reagents and Materials
Successful implementation of the cloning workflow requires specific, high-quality reagents. The following table details essential components for each stage of the process.
Table 3: Essential Research Reagents for Molecular Cloning
| Reagent Category | Specific Examples | Function | Application Notes |
|---|---|---|---|
| Restriction Enzymes | EcoRI, BamHI, NotI, KpnI | Sequence-specific DNA cleavage | Choose enzymes based on recognition sites, buffer compatibility, and temperature requirements |
| DNA Modifying Enzymes | T4 DNA Ligase, Alkaline Phosphatase, T4 DNA Polymerase | DNA joining and end-modification | Critical for ligation efficiency and preventing vector self-circularization |
| Cloning Vectors | pUC19, pBR322, Gateway vectors, BACs | DNA molecule for insert propagation | Select based on insert size, host system, and downstream applications |
| Competent Cells | DH5α, TOP10, BL21(DE3) | Host for plasmid propagation | Choose based on transformation efficiency, genotype features, and application needs |
| Selection Agents | Ampicillin, Kanamycin, Chloramphenicol | Selective pressure for transformants | Use appropriate concentration for plasmid and bacterial strain |
| DNA Extraction Kits | Plasmid miniprep, gel extraction kits | DNA purification from various sources | Essential for obtaining high-quality DNA for cloning and analysis |
| PCR Reagents | Taq polymerase, high-fidelity polymerases, dNTPs | DNA amplification | High-fidelity enzymes critical for accurate amplification of neural genes |
Applications in Neuroscience Research
Molecular cloning techniques serve as fundamental tools for diverse neuroscience applications:
- Functional Studies of Neural Genes: Cloning enables isolation and manipulation of genes encoding neuronal proteins, ion channels, and receptors for structure-function studies [22].
- Protein Expression: Recombinant DNA technology allows large-scale production of neural proteins for biochemical, structural, and pharmacological characterization [22] [75].
- Disease Modeling: Cloning of disease-associated neuronal genes facilitates creation of cellular and animal models of neurological disorders [22].
- Gene Therapy Development: Viral vectors created through molecular cloning approaches show promise for treating neurodegenerative disorders like Parkinson's and Alzheimer's disease [76].
Troubleshooting Guide
Common challenges in the cloning workflow and their solutions:
- Low transformation efficiency: Check competent cell quality, ensure proper heat shock/electroporation parameters, verify antibiotic concentration.
- High background (empty vectors): Optimize insert:vector ratio, verify vector dephosphorylation, ensure proper ligation conditions.
- No colonies: Verify antibiotic activity, check DNA quality, confirm competent cell viability, ensure selection marker compatibility.
- Incorrect clones: Verify restriction sites, sequence inserts fully, use high-fidelity enzymes for fragment generation.
The five-step cloning workflow provides a systematic framework for generating and analyzing recombinant DNA molecules. For neuroscience researchers, mastering these techniques enables sophisticated manipulation of neural genes and regulatory elements, advancing our understanding of nervous system function and dysfunction. As cloning technologies continue to evolve with methods like CRISPR-Cas9 and advanced DNA assembly techniques [22], the precision and efficiency of genetic engineering for neuroscience applications will continue to improve, opening new avenues for investigating the complexity of the nervous system and developing novel therapeutic strategies for neurological disorders.
Optimizing Primer Design and Restriction Enzyme Selection for Neural Targets
The study of the nervous system requires the precise manipulation of neural genes and cell types. Within the broader context of molecular cloning and recombinant DNA technology, two fundamental processes are critical for success: the design of DNA primers that accurately target neural genes and the selection of restriction enzymes that enable the correct assembly of genetic constructs. Optimizing these tools allows neuroscientists to create sophisticated models, such as lineage-specific reporters in human induced pluripotent stem cells (hiPSCs), to investigate neurodevelopment, neuronal function, and the mechanisms underlying neurological diseases [20] [77]. This protocol details streamlined methods for these key steps, integrating modern computational and enzymatic approaches to enhance the efficiency and reliability of genetic engineering in neuroscience research.
Computational Primer Design for Neural Targets
Algorithm Selection for Robust Primer Design
The design of high-quality primers is a critical first step in ensuring successful PCR amplification of neural targets. The choice of algorithm depends on the nature of the target sequences and the required specificity. For highly conserved neural genes, a conserved region approach is effective, whereas for polymorphic targets or those requiring amplification across species, a filtration-based method that allows limited degeneracy is more appropriate.
Table 1: Comparison of Primer Design Algorithms for Neural Targets
| Algorithm/ Tool | Core Methodology | Best For Neural Target Types | Key Advantages |
|---|---|---|---|
| SADDLE [78] | Simulated annealing to minimize primer-dimer formation | Large, highly multiplexed panels (e.g., for profiling many neuronal transcripts) | Optimizes large primer sets (192-768 primers); drastically reduces primer dimers |
| DeGenPrime [79] | Conserved region finding or filtration with degenerate bases | Phylogenetically conserved genes; polymorphic loci (e.g., neurotransmitter receptors) | Handles MSA inputs; manages degeneracy to expand target range |
| Thermodynamic Method [80] | Local alignment followed by thermodynamic interaction assessment | Highly divergent sequences or precise subtype discrimination | Maximizes specificity & sensitivity based on binding affinity |
A Detailed Protocol for Primer Design and Validation
The following workflow ensures the selection of specific and efficient primers for neural genes.
Step 1: Input and Align Target Sequences
- Identify the neural target gene (e.g., ALDH1L1 for astrocytes, NEUROG2 for neuronal progenitors) [77].
- Obtain genomic DNA sequences from databases like NCBI.
- Perform a Multiple Sequence Alignment (MSA) using a tool like MAFFT if designing primers for a gene family or across species [79].
Step 2: Select a Design Strategy and Generate Candidates
- For Conserved Targets: Use the conserved region approach. The software scans the MSA for regions with no degenerate nucleotides that are long enough to accommodate a primer (e.g., 18-30 bp) [79].
- For Variable Targets: Use the filtration approach. Specify a minimum amplicon size and allow limited degeneracy. The algorithm will generate all possible primers and then apply filters [79].
Step 3: Apply Filtration and Scoring Criteria
- Degeneracy: Disqualify primers with degenerate bases (N) or more than one 3-fold degenerate base. A maximum of two 2-fold degenerate bases is typically allowed [79].
- GC Content: Filter for primers with 40-60% GC content. Avoid runs of identical nucleotides and ensure no more than 3 G or C nucleotides at the 3' end (GC clamp) [79] [77].
- Melting Temperature (Tm): Calculate Tm using the nearest-neighbor method. For a set of primers, ensure Tm values are within a narrow range (e.g., 50-65°C with <2°C difference between forward and reverse) [79].
- Secondary Structure: Apply a penalty scoring system to rank primers. Penalize primers with a high likelihood of forming hairpins (triloops or tetraloops) or self-dimers with a Gibbs free energy (ΔG°) below -3 kcal/mol [79].
Step 4: Experimental Validation
- Synthesize the selected primers.
- Perform a T7 Endonuclease I (T7E1) Assay to empirically test the efficiency and specificity of the primers, especially when used with CRISPR/Cas9 components [77].
- Sequence the PCR products to confirm accurate amplification of the intended neural target.
Strategic Restriction Enzyme Selection for Cloning Neural Constructs
Principles of Enzyme Selection
The selection of appropriate restriction enzymes is paramount for the successful cloning of neural gene constructs into plasmid vectors. The primary goal is to excise the insert and linearize the vector with compatible ends for ligation, without cutting within the insert itself.
Table 2: Key Considerations for Restriction Enzyme Selection
| Factor | Consideration | Application to Neural Targets |
|---|---|---|
| Unique Restriction Sites [81] | The enzyme's recognition site must be unique and not present within your neural gene's coding sequence. | Verify the entire sequence of your neural gene (e.g., SOX1, OLIG2) for accidental restriction sites. |
| Compatible Ends [82] | Using two different enzymes creates directional cloning. Ends generated must be compatible for ligation. | Essential for inserting neural reporters in the correct orientation relative to the promoter. |
| Methylation Sensitivity [82] | Some enzymes (e.g., BclI) are sensitive to Dam/Dcm methylation in common E. coli strains. | If an enzyme fails to cut, transform the plasmid into a dam-/dcm- strain like JM110. |
| Buffer Compatibility [82] | In double digests, a buffer must be chosen where both enzymes retain >75% activity. | Use supplier-provided buffer compatibility charts to select the optimal buffer. |
| Star Activity [82] | Nonspecific cutting can occur at high glycerol concentrations (>5%) or prolonged incubation. | Keep the final glycerol concentration in the reaction mix low to maintain specificity. |
A Practical Protocol for Restriction Digestion
This protocol outlines the steps for a standard double digest, which is common when preparing an insert and vector for directional cloning.
Step 1: In Silico Selection of Restriction Enzymes
- Analyze the sequence of your neural gene insert (e.g., a tdTomato reporter) and the destination plasmid vector using sequence analysis software.
- Choose two different restriction enzymes with recognition sites that are unique in the plasmid's multiple cloning site (MCS) and absent from your neural gene insert [81]. This ensures directional cloning.
Step 2: Reaction Setup for a Double Digest
- If enzymes are compatible in a single buffer, assemble the reaction on ice as follows [82]:
- Nuclease-Free Water: to 20 µL final volume
- 10X Restriction Buffer: 2 µL
- Acetylated BSA (1 mg/ml): 2 µL
- DNA (∼1 µg): 1 µL
- Restriction Enzyme 1 (10 U): 1 µL
- Restriction Enzyme 2 (10 U): 1 µL
- If no single buffer provides >75% activity for both enzymes, a sequential digest is required [82].
- Perform the first digest in the optimal buffer for enzyme #1.
- Purify the DNA using a kit like the Wizard SV Gel and PCR Clean-Up System.
- Perform the second digest in the optimal buffer for enzyme #2.
Step 3: Execution and Cleanup
- Mix the components gently by pipetting and collect the contents at the bottom of the tube.
- Incubate at the optimal temperature for both enzymes (typically 37°C) for 1-2 hours. For enzymes with different optimal temperatures, incubate first at the lower temperature, then the higher, with a purification step in between if needed [82].
- Purify the digested DNA using a cleanup kit to remove enzymes, salts, and BSA before proceeding to ligation.
Integrated Workflow: Application in Neural Reporter Line Generation
The following diagram and protocol integrate primer design and restriction enzyme use into a cohesive workflow for generating a neural-specific reporter in hiPSCs using CRISPR/Cas9-assisted homologous recombination [77].
Protocol Overview:
- Targeting Vector Construction:
- Design Primers: Design primers to amplify ∼1 kb 5' and ∼1.5 kb 3' homology arms from the target neural gene (e.g., ALDH1L1) [77].
- Select Restriction Enzymes: Choose unique enzymes to clone these arms into a targeting vector containing your reporter (e.g., EGFP) and a floxed antibiotic resistance cassette.
- Assemble Vector: Use conventional cloning or DNA assembly methods (e.g., Gibson Assembly) to construct the final targeting vector.
CRISPR Component Preparation:
- Design sgRNAs: Design single-guide RNAs (sgRNAs) targeting a site near the stop codon of the neural gene to maximize homologous recombination efficiency [77].
- Clone sgRNAs: Clone annealed oligonucleotides into a Cas9 or Cas9n (D10A nickase) expression vector (e.g., pX335) using a restriction enzyme like BbsI [77].
HiPSC Transfection and Selection:
- Co-transfect the targeting vector and CRISPR/Cas9 plasmids into hiPSCs.
- Apply antibiotic selection to enrich for cells that have incorporated the targeting vector.
Screening and Validation:
- Pick individual clones and expand them.
- Screen by genomic PCR across the 5' and 3' junctions of the integrated reporter to identify correctly targeted clones.
- Confirm by DNA sequencing.
- Functionally validate the reporter by differentiating the hiPSCs and confirming reporter expression in the correct neural lineage (e.g., astrocytes for ALDH1L1) [77].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Neural Target Cloning and Editing
| Reagent / Tool | Function | Example Use Case |
|---|---|---|
| Restriction Enzymes [82] | Cut DNA at specific sequences to generate fragments with compatible ends for ligation. | Preparing the neural gene insert and linearizing the plasmid vector. |
| T4 DNA Ligase [82] | Joins DNA fragments with compatible ends by catalyzing phosphodiester bond formation. | Ligating the neural gene insert into the plasmid backbone. |
| DNA Polymerases [82] | Amplify DNA fragments in PCR. High-fidelity polymerases are used for cloning. | Amplifying homology arms or the neural gene coding sequence from genomic DNA or cDNA. |
| CRISPR/Cas9 System [77] [22] | Creates double-strand breaks in the genome at specified locations to stimulate homologous recombination. | Targeted integration of reporters into specific neural loci in hiPSCs. |
| Targeting Vectors [77] | Plasmid DNA containing the desired modification (e.g., reporter) flanked by homology arms. | Serving as the template for homologous recombination into the neural gene of interest. |
| HiPSC Lines [77] | A renewable source of human cells that can be differentiated into any neural cell type. | The cellular host for generating neural lineage-specific reporter lines. |
Molecular cloning is a foundational technique in neuroscience research, enabling the study of neural gene function, the production of neuroactive proteins, and the development of novel therapeutic agents. However, the path to successful cloning is often obstructed by technical challenges such as low plasmid yield, inefficient ligation, and failed bacterial transformation. These hurdles can significantly delay critical experiments in drug development and basic research. This application note provides a structured troubleshooting guide and optimized protocols to help researchers systematically overcome these common obstacles, ensuring efficient and reliable generation of recombinant DNA constructs for neurological applications.
Troubleshooting Common Cloning Hurdles
The following sections detail the primary causes and proven solutions for the most frequent cloning issues. A summary of these problems and their fixes can be found in Table 1.
Table 1: Comprehensive Troubleshooting Guide for Molecular Cloning
| Problem & Specific Symptoms | Root Cause | Recommended Solution |
|---|---|---|
| Few or No Transformants [83] [84] | ||
| No colonies on selective plates | Cells not viable or low transformation efficiency | Transform an uncut plasmid (e.g., pUC19) to check cell viability and efficiency. Use commercial high-efficiency competent cells (≥1x10⁹ CFU/µg) [83]. |
| Cells viable, but no transformants | DNA fragment is toxic to cells | Incubate plates at lower temperature (25–30°C). Use specialized strains (e.g., NEB-5-alpha F´ Iq) for tighter transcriptional control [83] [84]. |
| Inefficient ligation | Ensure at least one DNA fragment has a 5´ phosphate. Vary insert:vector molar ratio from 1:1 to 1:10. Use fresh ligation buffer (ATP degrades) [83] [85]. | |
| Construct too large | Use strains suited for large plasmids (e.g., NEB 10-beta, NEB Stable). Use electroporation for constructs >10 kb [83]. | |
| PEG in ligation mix (electroporation) | Clean up ligation mix with a PCR & DNA Cleanup Kit prior to electroporation [83]. | |
| Low Plasmid Yield [86] | ||
| Mini-prep yields <5 µg | Low copy number vector or large insert | Verify vector origin. For low/medium copy vectors, use more culture volume. For high copy, culture oversaturation can be an issue [86]. |
| Problematic (toxic/unstable) insert | Use specialized cell lines: STBL2 for unstable repeats, T7 Express LysY/Iq for toxic proteins [86]. | |
| Culture oversaturation or undergrowth | Grow culture to late log phase (OD600 ~3). Avoid overnight cultures from old colonies. Use a fresh starter culture [86]. | |
| Insufficient antibiotic pressure | Use fresh antibiotic stock. For relaxed origin plasmids (pMB1/ColE1), consider chloramphenicol amplification [86]. | |
| Inefficient bacterial lysis | For low copy plasmids, double resuspension/lysis/neutralization buffer volumes. Mix by inverting continuously for 3 minutes during lysis [86]. | |
| High Background (Wrong Construct) [83] [84] | ||
| Many colonies, but few contain insert | Vector self-ligation due to incomplete digestion or inefficient dephosphorylation | Gel-purify digested vector. Ensure complete dephosphorylation of vector and its inactivation/removal [83] [84]. |
| Restriction enzyme(s) didn’t cleave completely | Check methylation sensitivity. Clean up DNA to remove contaminants. Use recommended NEB buffers [83]. | |
| Antibiotic level too low or degraded | Confirm correct antibiotic concentration. Allow agar to cool before adding heat-sensitive antibiotics [84]. | |
| Colonies contain plasmid without insert | Satellite colonies from degraded antibiotic | Pick large, well-isolated colonies. Do not over-incubate plates (>16 hrs) [84]. |
| Inefficient Ligation [83] [85] | ||
| No ligated product on gel | Lack of 5' phosphate on insert (if from PCR) | Phosphorylate PCR products with T4 Polynucleotide Kinase (T4 PNK) prior to ligation [85]. |
| Incompatible DNA ends | Confirm ends are compatible. Use T4 DNA Ligase for blunt ends or single base-pair overhangs with PEG [83] [85]. | |
| Reaction inhibitors present | Clean up DNA prior to ligation to remove salts, EDTA, or proteins. Keep final glycerol concentration <5% [85]. |
Core Experimental Protocols
Protocol: Optimizing DNA Ligation
The ligation step, which joins the insert and vector, is critical for success. The following protocol is optimized for both sticky-end and blunt-end ligations [85].
Materials:
- T4 DNA Ligase and corresponding 10x Reaction Buffer (contains ATP)
- Vector DNA (20-100 ng per reaction)
- Insert DNA (calculated amount for desired molar ratio)
- 50% Polyethylene Glycol (PEG) 4000 solution (for blunt-end ligations only)
- Nuclease-free water
Method:
- Thaw reagents on ice. Gently vortex the 10x ligation buffer to mix and centrifuge briefly. Note: Buffer containing ATP and DTT is highly susceptible to freeze-thaw degradation; aliquot for single use.
- Set up the reaction on ice in a sterile microcentrifuge tube as shown in Table 2.
Table 2: Ligation Reaction Setup
| Component | Sticky-End Ligation | Blunt-End Ligation |
|---|---|---|
| Vector DNA | 20-100 ng | 20-100 ng |
| Insert DNA | x µL (see calculation below) | x µL (see calculation below) |
| 10x Ligation Buffer | 2 µL | 2 µL |
| 50% PEG 4000 | - | 2 µL |
| T4 DNA Ligase | 1.0-1.5 Weiss Units | 1.5-5.0 Weiss Units |
| Nuclease-free Water | to 20 µL final volume | to 20 µL final volume |
- Calculate Insert Mass: Use the following equation to calculate the mass (ng) of insert required for a desired molar ratio (e.g., 3:1 insert:vector) [85]:
ng of insert = (ng of vector × length of insert (bp)) / length of vector (bp) × desired molar ratioFor blunt-end ligations, a higher ratio (5:1 to 10:1) is recommended. - Incubate the reaction: Incubate at room temperature (22°C) for 10 minutes to 1 hour. Prolonged incubation is typically not necessary and can be detrimental for some applications.
- Transform competent cells: Use 1-5 µL of the ligation reaction to transform 50 µL of chemically competent cells. If using electrocompetent cells, it is essential to clean up the ligation reaction first to remove salts [83].
Protocol: High-Yield Plasmid Cultivation
Maximizing plasmid yield begins with healthy, high-density bacterial cultures [86].
Materials:
- LB broth and LB agar plates with appropriate antibiotic
- Fresh bacterial colony containing your plasmid
- Optional: Chloramphenicol stock solution (170 mg/mL in ethanol)
Method:
- Starter Culture: In the morning, inoculate 5 mL of LB (no antibiotic) with a fresh, well-isolated colony (from a plate no older than a few days). Grow with shaking at 37°C until the OD600 reaches ~1 (typically 4-6 hours).
- Refrigerate Starter: Place the starter culture in the refrigerator overnight. This synchronizes the cells and improves the efficiency of the main inoculation.
- Main Culture: The next morning, pellet the cells from the starter culture and resuspend in 1 mL of fresh, antibiotic-free LB. This step removes secreted beta-lactamase, which can degrade ampicillin.
- Dilution: Inoculate 100 mL of LB containing the appropriate antibiotic with 1 mL of the resuspended starter culture.
- Growth: Grow the culture with vigorous shaking at 37°C until the OD600 reaches approximately 3 (late log phase). Do not allow the culture to become oversaturated.
- Harvest: Pellet the cells by centrifugation. The cell pellet can be processed immediately for plasmid purification or stored at -20°C.
Optional Chloramphenicol Amplification: For plasmids with a relaxed origin (pMB1/ColE1, e.g., pUC, pGEM, pBR322 derivatives), culture density can be increased by adding 170 µg/mL of chloramphenicol to the main culture once it reaches saturation. Incubate for a further 16 hours. This halts protein synthesis and cell division but allows plasmid replication to continue, dramatically increasing copy number [86].
The Molecular Cloning Workflow
The following diagram outlines the key steps in a standard molecular cloning experiment, from planning to analysis, and highlights where common failures occur.
Figure 1: The molecular cloning workflow with key failure points identified. Success requires careful optimization at each step.
The Scientist's Toolkit: Essential Reagents & Strains
Selecting the correct enzymes and bacterial strains is fundamental to overcoming specific cloning hurdles.
Table 3: Essential Research Reagents and Strains for Cloning
| Item | Function & Key Characteristics | Example Use Cases |
|---|---|---|
| T4 DNA Ligase [85] | Joins DNA fragments by catalyzing phosphodiester bond formation. Requires 5'-phosphate and 3'-OH. | Standard sticky-end and blunt-end ligations. |
| High-Fidelity DNA Polymerase (e.g., Q5) [83] [87] | PCR amplification with extremely low error rate. Produces blunt-ended fragments. | Generating high-quality, mutation-free inserts for cloning. |
| T4 Polynucleotide Kinase (T4 PNK) [83] | Adds a 5' phosphate group to DNA fragments. Essential for ligating PCR products generated by proofreading polymerases. | Phosphorylating oligonucleotides or blunt-ended PCR products prior to ligation. |
| Alkaline Phosphatase (e.g., rSAP, CIP) [84] | Removes 5' phosphates from DNA to prevent vector self-ligation. Critical for reducing background. | Dephosphorylating a linearized vector backbone before ligation with an insert. |
| NEB 5-alpha Competent E. coli [83] | recA- endA- strain for high-efficiency transformation and stable propagation of high-copy plasmids. | General purpose cloning, plasmid propagation. |
| NEB Stable Competent E. coli [83] | Designed for stable propagation of "unstable" inserts (e.g., long repeats, toxic genes). | Cloning difficult DNA sequences that tend to recombine or delete in standard strains. |
| NEB 10-beta Competent E. coli [83] | mcrA- mcrBC- mrr- strain, incapable of degrading methylated DNA. | Cloning DNA from mammalian or plant sources, which often contains methylated cytosines. |
Successful molecular cloning in neuroscience research is achievable through meticulous planning, understanding the underlying biochemistry, and systematic troubleshooting. By implementing the protocols and solutions outlined here—such as optimizing ligation conditions, using high-yield cultivation methods, and selecting appropriate bacterial strains—researchers can effectively overcome the common hurdles of low yield, improper ligation, and failed transformation. This robust approach ensures the reliable production of high-quality DNA constructs, accelerating research into the complex mechanisms of the nervous system and the development of novel neurotherapeutics.
Best Practices for Screening and Selecting Transformed Cells with Neural Constructs
The integration of molecular cloning and recombinant DNA technology has become a cornerstone of modern neuroscience research, enabling the precise investigation of neural function and dysfunction. The ability to introduce genetic constructs into cells to express, inhibit, or modify neural targets is fundamental to modeling diseases, studying signaling pathways, and validating potential therapeutic targets. The critical step in this process is the efficient screening and selection of successfully transformed cells, ensuring that downstream experiments are conducted on a population accurately expressing the desired genetic modification. This protocol details best practices for this crucial phase, providing a robust framework for researchers and drug development professionals to generate reliable and reproducible data in neural contexts.
The Scientist's Toolkit: Essential Reagents for Screening and Selection
The following table catalogues the essential reagents required for the successful screening and selection of transformed neural cells.
Table 1: Key Research Reagent Solutions for Cell Screening and Selection
| Reagent / Tool | Function / Explanation in Screening and Selection |
|---|---|
| Restriction Enzymes | Molecular scissors used to linearize vector DNA prior to transfection and to confirm successful integration of the insert via diagnostic digest of isolated clones [82] [88]. |
| DNA Ligases | Enzymes that catalyze the joining of DNA fragments; crucial during vector construction to ligate the neural gene of interest into the plasmid backbone [82]. |
| Selection Antibiotics | Chemical agents (e.g., Puromycin, G418) added to culture media to eliminate untransformed cells. Only cells expressing the resistance gene on the vector survive [89] [88]. |
| Fluorescent Reporters | Genes like GFP or RFP encoded in the vector. Successfully transformed cells can be identified and isolated via fluorescence microscopy or Fluorescence-Activated Cell Sorting (FACS) [89] [88]. |
| CRISPR-Cas9 System | A genome-editing tool that, when coupled with a guide RNA (sgRNA) library, enables high-throughput genetic screening to identify genes affecting neural phenotypes [89]. |
| Polymerases | Enzymes like Taq polymerase are essential for Polymerase Chain Reaction (PCR), used to amplify and verify the presence of the inserted neural construct in candidate clones [88]. |
Experimental Workflow and Protocol
The process from transfection to a validated clonal population involves sequential stages of selection and analysis. The workflow is designed to maximize efficiency and confirmation of successful transformation.
Protocol 1: Initial Selection and Clonal Isolation
Objective: To eliminate untransformed cells and isolate single-cell derived clones for downstream analysis.
Materials:
- Cell culture of the neural cell line (e.g., SH-SY5Y, primary neurons, induced pluripotent stem cell-derived neurons)
- Recombinant DNA construct containing the neural gene of interest and a selection marker (e.g., antibiotic resistance, fluorescent protein)
- Appropriate transfection reagent (e.g., lipofectamine, calcium phosphate)
- Complete cell culture media
- Selection media containing the appropriate antibiotic (e.g., G418, Puromycin) or equipment for Fluorescence-Activated Cell Sorting (FACS)
Methodology:
- Transfection: Introduce the recombinant neural construct into the target cells using the optimized transfection method for your cell line. Include a negative control (no DNA or empty vector) to assess background survival.
- Recovery: Incubate the cells for 24-48 hours in complete growth media without selection to allow for vector integration and transgene expression.
- Initial Selection:
- Antibiotic Selection: Replace the media with selection media containing the predetermined optimal concentration of antibiotic. Change the media every 2-3 days to remove dead cells and maintain selection pressure. Non-transfected cells will begin to die off over 3-7 days.
- FACS Selection: For fluorescent reporters, harvest the cells and resuspend in a FACS-compatible buffer. Use a flow cytometer to sort the top 5-10% of fluorescent cells into a new culture vessel.
- Clonal Isolation: Once resistant or fluorescent pools are established, dissociate the cells to a single-cell suspension. Seed the cells at a very low density (e.g., 1-10 cells per well) in a 96-well plate to allow for the growth of discrete monoclonal colonies. Alternatively, use FACS to deposit a single cell into each well of a 96-well plate.
- Expansion: Monitor the plates and allow individual clones to expand for 1-2 weeks, with careful media changes, until sufficient cells are available for validation.
Protocol 2: Molecular Validation of Clones
Objective: To confirm the successful integration and presence of the neural construct within the genomic DNA of the isolated clonal populations.
Materials:
- Genomic DNA extraction kit
- PCR reagents: Taq polymerase, dNTPs, primers specific for your neural construct
- Gel electrophoresis equipment
- Restriction enzymes and appropriate buffer [82]
- Agarose gel
Methodology:
- Genomic DNA Extraction: Harvest a portion of the cells from each expanding clone and extract genomic DNA using a commercial kit.
- PCR Screening:
- Design primers that flank the insertion site of your neural construct or are specific to the transgene sequence.
- Set up PCR reactions using the extracted genomic DNA as a template.
- Run the PCR products on an agarose gel. Clones containing the construct will show a band of the expected size, while negative controls will not.
- Restriction Digest Analysis:
- Use the isolated genomic DNA or a miniprepped plasmid if the construct is maintained episomally.
- Perform a diagnostic restriction digest using enzymes that cut at specific sites within your construct to release a fragment of a known, predictable size [82].
- Analyze the digest fragments on an agarose gel to confirm the correct molecular architecture of the integrated construct.
Protocol 3: Functional Validation via Advanced Screening (e.g., CRISPR-StAR)
Objective: To assess the functional consequence of the genetic perturbation in a complex, physiologically relevant context, controlling for heterogeneity.
Background: Conventional screening in complex models like neural organoids or in vivo environments is confounded by bottleneck effects and clonal diversity. The CRISPR-StAR (Stochastic Activation by Recombination) method overcomes this by generating internal controls within each single-cell-derived clone, enabling high-resolution genetic screening [89].
Materials:
- CRISPR-StAR vector library containing inducible sgRNAs
- Target neural cells expressing Cas9 and Cre::ERT2
- Tamoxifen or 4-OHT for induction
- Unique Molecular Identifier (UMI) barcoding system
- Next-generation sequencing platform
Methodology:
- Library Transduction: Transduce the neural cell population with the CRISPR-StAR sgRNA library at a high multiplicity of infection (MOI) to ensure good coverage.
- Engraftment & Clonal Expansion: Transplant the transduced cells into your complex model (e.g., animal brain, organoid) and allow them to engraft and form single-cell-derived clones tracked by UMIs.
- Induction of Recombination: Administer tamoxifen to activate Cre::ERT2. This stochastically and irreversibly generates two populations within each UMI-marked clone: one with the sgRNA in an active state and the other with the sgRNA in an inactive state, serving as an internal control [89].
- Phenotypic Screening and Analysis: After a period for phenotypic expression (e.g., 14 days), harvest the cells and quantify the abundance of active vs. inactive sgRNAs within each UMI clone via next-generation sequencing.
- Data Interpretation: A depletion or enrichment of active sgRNAs in a specific UMI population, relative to its own internal control, indicates a gene-dependent effect on survival or proliferation under the screening conditions. This internal control negates noise from clonal heterogeneity [89].
Table 2: Quantitative Performance Comparison of Screening Methods
| Method | Key Metric | Performance / Value | Key Advantage |
|---|---|---|---|
| Conventional CRISPR Screening | Reproducibility (Pearson R) at low coverage [89] | ~0.07 | Standardized, widely accessible. |
| CRISPR-StAR Screening | Reproducibility (Pearson R) at low coverage [89] | >0.68 | Superior accuracy in complex, heterogeneous models. |
| Fluorescence-Based Sorting | Purity of isolated population | >95% | High-purity isolation of live, expressing cells. |
| Antibiotic Selection | Time to establish stable pool | 1-3 weeks | Cost-effective for large populations; does not require specialized equipment. |
The rigorous screening and selection of transformed neural cells is a critical determinant of success in neuroscience research. By employing a combination of selective pressure, molecular verification, and advanced functional screening methods like CRISPR-StAR, researchers can ensure the integrity of their experimental models. The protocols outlined here, from basic antibiotic selection to sophisticated internally controlled screening, provide a comprehensive roadmap for generating high-quality, reliable data. This structured approach ultimately accelerates the pace of discovery in understanding neural mechanisms and developing novel therapeutics for neurological disorders.
Leveraging Automated Plasmid Purification and Commercial Gene Services for Efficiency
In the field of neuroscience research, the demand for high-quality plasmid DNA (pDNA) for applications such as neuronal transfection, viral vector production, and gene therapy development is growing rapidly. Molecular cloning and recombinant DNA technology form the backbone of these endeavors, enabling the study of neural circuits, disease mechanisms, and potential therapeutic interventions [15] [20]. However, traditional manual methods for plasmid purification are often labor-intensive, time-consuming, and prone to variability, creating significant bottlenecks in research pipelines [90] [91].
This application note details how leveraging automated plasmid purification and commercial gene synthesis services can significantly enhance efficiency and reproducibility. We provide structured quantitative data, detailed protocols, and a curated toolkit to help neuroscience researchers and drug development professionals streamline their molecular cloning workflows, thereby accelerating the pace of discovery in genetic neurological diseases [92] [15].
Application Note: Evaluating Automated Plasmid Purification Systems
The transition to automation in plasmid preparation addresses critical limitations of manual methods, notably by reducing hands-on time and improving process consistency. This is particularly vital in neuroscience where experiments often require high-quality, transfection-grade DNA for sensitive primary neuronal cultures or the production of adeno-associated viral (AAV) vectors.
Performance Comparison of Purification Methods
The following table summarizes the key operational and output metrics for two automated platforms compared to a traditional manual method.
Table 1: Quantitative Comparison of Plasmid Purification Methods
| Parameter | Manual Column-Based (Midi Prep) | KingFisher PlasmidPro Maxi (Automated) | Lynx with MidiPure IMCStips (Automated) |
|---|---|---|---|
| Scale | Midi | Maxi | Midi to Maxi |
| Average Hands-on Time | ~60 minutes | <5 minutes [90] | Significant reduction [93] |
| Total Process Time | ~90 minutes | ~75 minutes [90] | Not Specified |
| Typical Yield | 251 ± 9 µg (Benchmark) | Consistent high yield [90] | 233 ± 3 µg (with 2 tips) [93] |
| Purity (A260/A280) | Optimal range | Optimal ratios (e.g., ~1.8) [90] | Consistently in optimal range [93] |
| % Supercoiled DNA | ~88% (for 6.6 kb plasmid) | >80% (across 3 plasmid sizes) [90] | Confirmed via TapeStation [93] |
| Suitable Downstream Applications | Transfection, cloning | Gene therapy, IVT mRNA synthesis, transfection [90] | Gene therapy, cloning, sequencing [93] |
Key Advantages for Neuroscience Research
- Workflow Efficiency: Automated systems drastically reduce hands-on time. For instance, the KingFisher PlasmidPro workflow requires less than 5 minutes of hands-on time, freeing researchers for data analysis or other experimental tasks [90].
- Enhanced Reproducibility: Standardized protocols and pre-filled reagent cartridges minimize user-induced variability, ensuring consistent pDNA quality crucial for longitudinal neuronal culture studies [90].
- Scalability for Pre-clinical Research: These systems enable the reliable production of large quantities of high-quality pDNA, which is essential for pre-clinical development of gene therapies for neurological disorders like Huntington's disease [90] [92].
Protocol: Automated Plasmid DNA Extraction fromE. coli
This protocol is adapted from a publicly available method [94] for automated extraction of plasmid DNA from 24 E. coli cultures using a liquid handler, optimized for neuroscience research applications.
Research Reagent Solutions
Table 2: Essential Materials for Automated Plasmid Extraction
| Item | Function/Description |
|---|---|
| Hamilton STAR Liquid Handler | Automated liquid handling system with 8- and 96-channel pipetting heads and liquid-level sensing. |
| Positive Pressure Filter Press (e.g., Hamilton MPE2) | Drives solutions through filter and binding plates without centrifugation. |
| QIAprep Spin Miniprep Kit Reagents | Provides resuspension, lysis, neutralization, and binding buffers. |
| Nuclease-free Water | Elution of purified plasmid DNA, ensuring stability for long-term storage. |
| 96-well Deep Well Plate | Culturing and processing of bacterial samples in a high-throughput format. |
| 96-well Glass Fiber Binding Plate | Silica-based solid matrix for selective binding of DNA during purification. |
| Wide Bore Pipette Tips | Prevent shearing of high-molecular-weight DNA during mixing and transfer. |
Detailed Methodology
Cell Pellet Preparation
- Pellet 1.7 mL of overnight E. coli cultures in a 96-well deep-well plate by centrifugation at 3878 g for 10 minutes at 23°C.
- Using the liquid handler's 8-channel head, carefully remove and discard the supernatant without disturbing the cell pellet.
Cell Resuspension and Lysis
- Add 200 µL of Resuspension Buffer (P1 containing RNase A) to each well and mix 10 times by repeated aspiration and dispensing via the 8-channel head.
- Transfer the resuspended cells to a new 96-well plate on a microplate shaker.
- Add 250 µL of Lysate Buffer (P2) to each sample using the 96-channel head. Mix by shaking the plate at 90 rpm for 2 minutes. Do not vortex, as this will cause genomic DNA contamination.
Neutralization and Filtration
- Add 350 µL of cold (4°C) Neutralization Buffer (N3) to each sample using the 96-channel head. Mix by shaking at 90 rpm for 2 minutes. A white precipitate will form.
- Using wide-bore tips, gently mix the lysate with 3 cycles of aspiration and dispensing. Transfer the mixture to a 96-well filter plate and let it settle for 2 minutes.
- Use the positive pressure filter press to push the cleared lysate through the filter plate into a new deep-well plate (20 psi for 240 s, then 65 psi for 60 s).
DNA Binding and Washing
- Transfer the cleared lysate to a 96-well glass fiber binding plate. Push the solution through the binding plate using the filter press at 40 psi for 60 s.
- Add 900 µL of Binding Buffer (PB) to each well and push through at 40 psi for 60 s.
- Add 900 µL of Wash Buffer (PE) to each well and push through at 40 psi for 60 s. Dry the binding plate by increasing pressure to 65 psi for 420 s.
DNA Elution
- Place the dried binding plate on top of a new, low-binding 96-well elution plate.
- Add 130 µL of nuclease-free water (pre-warmed to 60°C) to each well using the 8-channel head, jet-dispensing directly onto the filter.
- Wait for 5 minutes to allow DNA to hydrate. Elute the DNA into the elution plate using the filter press at 65 psi for 7 minutes.
- Seal the plate and store the purified plasmid DNA at 4°C for immediate use or -20°C for long-term storage.
Workflow Visualization
The following diagram illustrates the automated plasmid purification protocol workflow:
Integration with Commercial Gene Services
For maximum efficiency, automated plasmid purification should be integrated with commercial gene synthesis and cloning services. This combination allows researchers to bypass the initial, often time-consuming steps of gene cloning and vector construction.
- Accelerated Construct Generation: Services like Thermo Fisher's GeneArt Gene and Protein Synthesis Services provide synthesized genes, cloned vectors, and even fully sequenced plasmids, moving directly from sequence to functional DNA [90]. This is invaluable for rapidly testing hypotheses about gene function in neuronal models.
- Focus on Core Research: Outsourcing gene synthesis enables neuroscience labs to dedicate more resources to downstream applications—such as functional assays in primary neurons or animal models—rather than spending weeks on molecular cloning [90] [95].
- Quality Assurance: Reputable service providers deliver sequence-verified constructs, ensuring the integrity of genetic material used in critical experiments, which is foundational for reproducible research in complex fields like neurogenetics [90].
The integration of automated plasmid purification and commercial gene services represents a significant strategic advancement for molecular cloning in neuroscience research. This approach directly addresses the pressing need for scalability, reproducibility, and efficiency in the development of cell and gene therapies, a market projected to reach $25.37 billion by 2025 [92]. The consistent, high-quality pDNA output is essential for sensitive downstream applications like producing viral vectors for neuronal gene delivery or mRNA for therapeutic synthesis [90].
Future advancements will likely involve greater integration of artificial intelligence for process optimization and the continued push towards standardization across the industry [92]. For neuroscience researchers, adopting these technologies is not merely a matter of convenience but a critical step in accelerating the translation of basic genetic discoveries into novel therapeutic strategies for debilitating neurological diseases.
Confirming and Comparing: Ensuring Accuracy and Selecting Optimal Cloning Strategies
In the field of neuroscience research, the precise analysis of gene function and expression is paramount for understanding neuronal development, synaptic plasticity, and the mechanisms underlying neurodegenerative diseases. Molecular cloning and recombinant DNA technology serve as foundational techniques for these investigations, often involving the creation of overexpression vectors, luciferase reporter constructs, and other plasmid-based tools to probe neural function [21]. A critical final step in many of these experimental pipelines is the validation of constructed plasmids and the analysis of genetic variants through DNA sequencing. For decades, Sanger sequencing, developed by Frederick Sanger in 1977, has been regarded as the undisputed gold standard for confirming DNA sequences due to its proven reliability and accuracy [96] [97]. This application note details robust protocols for the sequencing of PCR products and the analysis of resultant data, framing them within the context of a modern neuroscience research laboratory where validation confidence is non-negotiable.
The emergence of Next-Generation Sequencing (NGS) technologies, which provide high-throughput, massively parallel sequencing capabilities, has transformed genomic studies [96]. While NGS is increasingly used for discovery-based profiling, the validation of its findings, particularly for critical variants in neuronal genes, has traditionally relied upon Sanger sequencing. However, a pivotal study by researchers at the National Human Genome Research Institute (NHGRI) has challenged this practice, suggesting that NGS is as accurate—and potentially more accurate—than Sanger sequencing for validating certain types of variants [97]. This guide will therefore cover validation strategies employing both technologies, providing neuroscientists with a comprehensive toolkit for ensuring data integrity in their molecular cloning workflows.
Sequencing Technologies for Validation
The choice of sequencing technology is determined by the experimental goal. Sanger sequencing is ideal for targeted confirmation, such as verifying the sequence of a cloned insert in a plasmid or confirming a specific mutation in a neuronal gene model. In contrast, NGS is suited for broader discovery, like identifying unknown transcriptional variants or profiling entire gene families involved in a neuropathological process.
Sanger Sequencing
Sanger sequencing, or the chain-termination method, is a technique based on the selective incorporation of fluorescently labeled, chain-terminating dideoxynucleotides (ddNTPs) during in vitro DNA replication [96]. The resulting DNA fragments are separated by capillary electrophoresis, and the sequence is determined by detecting the fluorescent signal of each terminal nucleotide. Its high accuracy for reads up to ~1000 base pairs makes it exceptionally well-suited for validating cloned constructs, PCR products, and targeted genomic regions [96] [82].
Next-Generation Sequencing (NGS)
NGS refers to a suite of high-throughput technologies that perform massively parallel sequencing, enabling the simultaneous determination of millions to billions of DNA fragments [96]. Unlike Sanger, which requires the physical separation of sequencing reactions, NGS reactions occur on a solid surface (e.g., glass flow cells or beads), where they are spatially separated and amplified to form clusters, and then sequenced in parallel [96]. Common NGS platforms, such as those from Illumina, utilize a sequencing-by-synthesis (SBS) approach with reversibly terminated fluorescent nucleotides [96]. The tremendous depth of coverage provided by NGS allows for the sensitive detection of low-frequency variants, a feature less accessible to traditional Sanger sequencing.
Comparative Analysis of Sequencing Technologies
The following table summarizes the core technical and performance characteristics of Sanger and Next-Generation Sequencing methodologies, highlighting their distinct roles in a research pipeline.
Table 1: Comparison of Sanger and Next-Generation Sequencing (NGS) Technologies
| Feature | Sanger Sequencing | Next-Generation Sequencing (NGS) |
|---|---|---|
| Underlying Principle | Dideoxy chain-termination with fluorescent labels [96] | Massively parallel sequencing-by-synthesis [96] |
| Throughput | Low (typically 1-384 samples per run) [97] | Very High (millions to billions of reads per run) [96] [97] |
| Read Length | Long (up to 1000 bp) [96] | Short (50-300 bp, platform-dependent) [96] |
| Typical Applications | Validation of clones, PCR products, and targeted mutations; confirmation of NGS findings [96] [82] | Whole genome, exome, and transcriptome sequencing; variant discovery; metagenomics [96] |
| Key Strength | High accuracy for individual, targeted sequences; considered the historical "gold standard" [96] [97] | Unbiased, high-throughput discovery; capable of detecting low-frequency variants [96] |
| Primary Limitation | Low throughput and high cost per base for large-scale projects [96] | Higher initial instrument cost; complex data analysis and storage requirements [96] |
| Cost per Sample | Higher for low-plexity targets | Lower for high-plexity targets |
A landmark study from the NHGRI directly compared the validation accuracy of these two methods. The researchers analyzed millions of DNA base pairs from ClinSeq participants that had been sequenced by both NGS and Sanger. In a comparison of 5,660 variants identified by NGS, only 19 were not initially confirmed by Sanger. Upon repeat Sanger sequencing, 17 of these 19 discrepancies were found to be errors in the initial Sanger read, not the NGS call. This resulted in a NGS accuracy of 99.965% and suggested that routine Sanger confirmation of NGS variants may introduce more errors than it corrects for single base substitutions [97].
Experimental Protocols
Protocol 1: Sanger Sequencing of PCR Products
This protocol describes the process from PCR amplification to sequence analysis, which can be applied to validate constructs like neuronal overexpression plasmids.
PCR Amplification and Purification:
- Amplify the target DNA fragment from your sample (e.g., purified plasmid, cDNA from neuronal culture) using a high-fidelity DNA polymerase.
- Purify the PCR product to remove primers, nucleotides, and enzymes. Use a commercial PCR purification kit or perform gel extraction if non-specific products are present [98] [82].
Cycle Sequencing Reaction:
- Set up the sequencing reaction using 5-20 ng of purified PCR product per 100 base pairs of sequence length.
- A typical reaction mix includes:
- PCR Product: 1-3 µL (10-100 ng total)
- Sequencing Primer: 1 µL (3.2 pmol/µL)
- Sequencing Reaction Mix (BigDye Terminator, etc.): 2-4 µL
- Nuclease-Free Water: to 10-20 µL final volume
- Cycling Conditions:
- 96°C for 1 minute (initial denaturation)
- 25 cycles of: 96°C for 10 seconds, 50°C for 5 seconds, 60°C for 4 minutes.
Post-Reaction Purification:
- Purify the extension products to remove unincorporated dye terminators. This can be done using ethanol/sodium acetate precipitation or commercial column-based kits.
Capillary Electrophoresis:
- Resuspend the purified product in a formamide-based buffer and denature.
- Load onto a capillary electrophoresis sequencer. The instrument will separate fragments by size and detect the fluorescent dye associated with each terminal ddNTP.
Data Analysis:
- The sequencer's software will generate a chromatogram (electropherogram) representing the DNA sequence.
- Analyze the chromatogram using sequence analysis software (e.g., Geneious, Sequencher). Compare the experimental sequence to the known reference sequence to identify any variations [96].
Protocol 2: Validating NGS-Derived Variants
This protocol outlines a strategic approach for confirming genetic variants identified through NGS, which is critical for high-impact findings in candidate neuronal genes.
Variant Calling from NGS Data:
- Process raw NGS data through a standard bioinformatics pipeline, including alignment to a reference genome (e.g., GRCh38) and variant calling using tools like GATK or FreeBayes [96].
- Filter variants based on quality metrics such as read depth, mapping quality, and allele frequency to generate a high-confidence list of candidate variants.
Confirmation Strategy:
- For a small number of variants: Design PCR primers that flank the variant of interest, typically generating an amplicon of 300-500 bp. Proceed with Sanger sequencing of the PCR product as described in Protocol 1. The decision to use Sanger validation should be based on the clinical or biological significance of the variant, not as an automatic routine [97].
- For a larger number of variants: Consider using a targeted NGS approach, such as designing a custom panel to re-sequence all candidate variants and their genomic context. This can be more cost-effective than Sanger for larger variant sets.
Data Interpretation:
- For Sanger validation, visually inspect the chromatogram at the genomic coordinate of the candidate variant to confirm its presence.
- Classify the confirmed variant using established guidelines (e.g., ACMG) as Pathogenic, Likely Pathogenic, Variant of Uncertain Significance (VUS), Likely Benign, or Benign [96].
The Scientist's Toolkit: Research Reagent Solutions
The following table lists essential reagents and materials required for the sequencing and validation workflows described in this note.
Table 2: Essential Research Reagents for Sequencing and Validation
| Reagent/Material | Function/Application |
|---|---|
| High-Fidelity DNA Polymerase | Amplification of target DNA for sequencing with superior accuracy to minimize PCR-introduced errors [82]. |
| Cycle Sequencing Kit (e.g., BigDye) | Contains enzymes and fluorescently labeled ddNTPs for the Sanger chain-termination sequencing reaction. |
| PCR Purification Kit | Removal of primers, salts, and enzymes from PCR products prior to sequencing. |
| Sanger Sequencing Primers | Gene-specific or universal primers (M13 forward/reverse) designed to initiate sequencing adjacent to the target region. |
| Agarose | Gel electrophoresis to size-select and purify specific PCR amplicons. |
| Cloning Vector (e.g., TA Vector) | For direct cloning of PCR products, facilitating sequencing and archiving of specific alleles [98]. |
| Competent E. coli Cells | Bacterial transformation to amplify plasmid DNA for sequencing or storage [98] [21]. |
| Plasmid Purification Kit | Isolation of high-quality plasmid DNA from bacterial cultures for use as a sequencing template [21]. |
| NGS Library Prep Kit | Platform-specific reagents for fragmenting DNA, attaching adapters, and preparing libraries for massively parallel sequencing [96]. |
Workflow Visualization
The following diagram illustrates the integrated decision-making process and experimental workflow for validating DNA sequences using Sanger and NGS methods.
The paradigm for gold-standard validation in DNA sequencing is evolving. While Sanger sequencing remains a highly reliable and accessible method for confirming specific sequences, such as cloned genes in neuronal expression vectors, contemporary evidence demonstrates that NGS possesses a level of accuracy that challenges the need for routine Sanger confirmation of all NGS-derived variants [97]. The optimal validation strategy in neuroscience research is therefore context-dependent. Researchers should leverage the high-throughput power of NGS for discovery and apply targeted Sanger sequencing judiciously, based on the biological significance of the finding and the required level of confidence for publication or clinical application. This nuanced approach ensures both rigor and efficiency in the molecular validation workflows that underpin advanced neuroscience research.
In the field of neuroscience research, understanding the precise regulation of gene expression is paramount for unraveling the molecular mechanisms underlying brain function, development, and disease. Molecular cloning and recombinant DNA technologies provide the foundational tools for these investigations, enabling researchers to dissect complex genetic programs within the brain's highly heterogeneous cellular environments. Two complementary methodologies have become cornerstone techniques for confirming and quantifying gene expression: reporter gene assays and RNA sequencing (RNA-seq). Reporter genes offer a direct, functional readout of transcriptional activity from specific genetic regulatory elements, while RNA-seq provides a comprehensive, unbiased profile of the entire transcriptome. This application note details integrated protocols employing these powerful techniques, framed within the context of contemporary neuroscience research challenges, including the characterization of neural signaling pathways, the validation of disease-associated genetic variants, and the identification of novel therapeutic targets for neuropsychiatric disorders [99]. The combination of these approaches allows for both targeted hypothesis testing and discovery-driven exploration, offering a robust framework for validating bioinformatic predictions and generating mechanistic insights into brain function and dysfunction.
Methodologies and Experimental Protocols
Reporter Gene Assay for Promoter/Enhancer Activity
Reporter gene assays are a venerable tool for studying signaling pathways and inferring the activity of pathway-specific transcription factors [100]. The following protocol describes a method to investigate the function of a putative neural enhancer element (e.g., one identified via GWAS to be associated with a neuropsychiatric disorder) by cloning it into a reporter vector and measuring its activity in a relevant neural cell model.
Protocol Steps:
- Vector Design and Cloning: Clone the genomic sequence of the putative enhancer (typically 200-1000 bp) into a multiple cloning site upstream of a minimal promoter driving a reporter gene in a lentiviral vector. The most common reporter genes are luciferases (firefly, NanoLuc) or fluorescent proteins (GFP, mCherry) [101] [102]. A key design feature is the inclusion of a unique sequence-tag within the 3' UTR of the reporter gene, which enables the tracking of the transcriptional output from that specific vector in a pooled format via RNA-seq [100].
- Cell Transduction: Transduce a relevant neural cell type (e.g., primary neurons, induced pluripotent stem cell (iPSC)-derived neural progenitors, or a neuronal cell line) with the lentiviral reporter vector. Include a control vector (e.g., with a scrambled sequence or minimal promoter only) to establish baseline activity. For normalization in dual-reporter assays, a second control vector expressing a different reporter (e.g., Renilla luciferase) under a constitutive promoter can be co-transduced [102].
- Stimulation and Incubation: Incubate the transduced cells for 24-48 hours. Apply relevant stimuli or inhibitors to probe specific signaling pathways (e.g., BDNF to activate MAPK signaling, or a small molecule to investigate a drug's mechanism of action) [100].
- Luminescence Measurement and Analysis: Lyse the cells and assay for reporter activity. For luciferase reporters, add the appropriate substrate (e.g., D-luciferin for firefly luciferase, furimazine for NanoLuc) and measure luminescence intensity with a microplate reader [101]. Normalize the experimental reporter activity to the control reporter signal to account for variations in cell viability and transduction efficiency.
Table 1: Common Bioluminescent Reporter Genes and Their Characteristics
| Reporter Gene | Source | Size | Substrate | Emission | Key Features |
|---|---|---|---|---|---|
| Firefly Luciferase | Photinus pyralis | 61 kDa | D-luciferin + ATP | 550-570 nm (Yellow-green) | High sensitivity, glow-type reaction with CoA [101] |
| NanoLuc Luciferase | Oplophorus gracilirostris | 19.1 kDa | Furimazine | 465 nm (Blue) | Small size, high intensity, ATP-independent, >2h signal half-life [101] |
| Renilla Luciferase | Renilla reniformis | 36 kDa | Coelenterazine | 480 nm (Blue) | Useful as a normalizing control in dual-assay systems [101] |
Parallel Reporter Assay with Integrated RNA-seq (TF-seq)
To overcome the throughput limitations of traditional reporter assays and capture complex signaling dynamics, parallel reporter assays like Transcription Factor activity sequencing (TF-seq) can be employed. TF-seq enables the parallel measurement of more than 40 signaling pathway activities alongside the global transcriptome from the same sample [100].
Protocol Steps:
- Pooled Reporter Library Preparation: A complex pool of 58 distinct lentiviral reporter vectors is used. Each vector contains a unique transcription factor response element (RE) and a corresponding unique sequence-tag (RE-tag) in the 3' UTR of the reporter gene [100].
- Cell Transduction and Stimulation: Transduce the cell population of interest (e.g., primary macrophages or iPSC-derived microglia) with the entire pooled library of viral particles. After transduction, stimulate the cells with the desired agent (e.g., a microbial stimulus or small molecule) [100].
- Parallel RNA-seq Library Preparation: Lyse the transduced cells directly in the culture plate. Perform reverse transcription using primers specific to the reporter gene and poly-dT primers for the global transcriptome. These primers are barcoded with well-specific tags and Unique Molecular Identifiers (UMIs) to enable sample multiplexing and accurate digital counting of RNA molecules [100].
- Sequencing and Data Analysis: Separately amplify and sequence the two cDNA pools: the RE-tagged reporter amplicons and the global 3' digital gene expression (3' DGE) library. In the resulting sequencing data, the RE-tag counts are used to infer the activity of each specific pathway, while the 3' DGE library provides standard global transcriptomic data. This allows for direct integration of pathway activity inferences with transcriptional changes [100].
The following workflow diagram illustrates the key steps in the TF-seq protocol:
Single-Cell RNA Sequencing (scRNA-seq) for Neural Cell Typing
The cellular heterogeneity of the brain makes single-cell RNA sequencing (scRNA-seq) an invaluable tool for neuroscience. It allows for the classification of diverse neuronal and glial subtypes and the detection of cell-type-specific molecular changes in disease [103] [99].
Protocol Steps:
- Single-Cell/Nucleus Suspension: Prepare a single-cell or single-nucleus suspension from fresh or frozen neural tissue. For brain tissue, single-nucleus RNA-seq (snRNA-seq) is often preferred due to the ease of isolation, better preservation of neuronal RNA, and reduced cell-type bias compared to whole-cell dissociation [99].
- Library Generation: Load the suspension onto a microfluidic device (e.g., 10X Chromium) to encapsulate individual cells/nuclei into droplets with barcoded beads. Within each droplet, cell lysis occurs, and mRNA is reverse-transcribed into cDNA with cell-specific barcodes and UMIs [103].
- Sequencing and Computational Analysis: Sequence the libraries using standard NGS platforms. Pre-process the data using pipelines like Cell Ranger to generate a cell-by-gene matrix. Downstream analysis includes dimensionality reduction (UMAP/t-SNE), unsupervised clustering to identify cell populations, and differential expression analysis to identify marker genes for each cluster [103].
Table 2: Comparison of Bulk and Single-Cell RNA-seq in Neuroscience Research
| Feature | Bulk RNA-seq | Single-Cell/Nucleus RNA-seq |
|---|---|---|
| Resolution | Average expression across all cells in a sample | Gene expression per individual cell |
| Best For | Identifying major transcriptional shifts; cost-effective profiling | Deconvolving cellular heterogeneity; identifying rare cell types; building cell atlases |
| Neuroscience Application | Analyzing homogenized brain regions | Classifying neuronal subtypes; tracing developmental trajectories; studying microglia states in disease |
| Key Consideration | Obscures cell-type-specific changes | Higher cost and computational burden; potential stress-induced artifacts from tissue dissociation [99] |
Results and Data Interpretation
Integrating Reporter Assay and RNA-seq Data
The power of the TF-seq approach lies in the simultaneous capture of two data modalities from the same biological sample. The results can be interpreted in two layers:
- Pathway Activity Inference: The count of each RE-tag from the sequencing data serves as a direct measure of the activity of the transcription factor and its upstream signaling pathway. For example, in a study of innate immune response in macrophages, TF-seq successfully captured dynamic activity changes in pathways like NFκB and STAT1 upon stimulation, which were not fully recapitulated by RNA-seq data alone [100].
- Global Transcriptomic Profiling: The standard RNA-seq data reveals all the gene expression changes occurring in the cell population under the experimental condition.
Integrating these datasets provides mechanistic insight. One can directly test whether the pathway activities inferred from the reporter assay are consistent with the observed changes in the expression of known target genes from the RNA-seq data. Furthermore, this integration can reveal unexpected signaling events. For instance, when investigating the anti-inflammatory natural product halofuginone, TF-seq identified an unexpected activation of NFκB alongside the suppression of STAT1, providing a more nuanced understanding of its mechanism of action [100].
Advanced Applications: Multi-Omic Single-Cell Sequencing
Emerging technologies are further enhancing our ability to link genotype to phenotype in complex neural systems. Single-cell DNA–RNA sequencing (SDR-seq) is a powerful new method that simultaneously profiles targeted genomic DNA loci and gene expression in thousands of single cells [104]. This allows for the direct association of coding and noncoding genetic variants—including those identified in neuropsychiatric GWAS—with distinct gene expression changes in their endogenous context, all while accounting for the cellular heterogeneity of the brain. This is a significant advance over traditional methods, which struggle to confidently link precise genotypes to cellular phenotypes in a pooled format.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Kits for Functional Gene Expression Analysis
| Item | Function | Example Application |
|---|---|---|
| Luciferase Reporter Vectors | Cloning backbone for regulatory elements; provides luminescent readout of transcriptional activity. | Testing promoter/enhancer function of neuropsychiatric risk variants [105] [102]. |
| NanoLuc Luciferase Assay | Detection reagent for NanoLuc reporter; provides high-intensity, glow-type luminescence. | High-sensitivity reporter assays in miniaturized formats (e.g., 384-well plates) for drug screening [101]. |
| Dual-Luciferase Reporter Assay | System to sequentially measure firefly and Renilla luciferase activity from a single sample. | Normalizing experimental reporter activity to a control reporter for data standardization [102]. |
| Lentiviral Packaging Systems | Production of lentiviral particles for efficient gene delivery into hard-to-transfect cells, like primary neurons. | Delivering reporter constructs or CRISPR-based perturbation tools into iPSC-derived neurons [100]. |
| Single-Cell RNA-seq Kits | (e.g., 10X Chromium) For generating barcoded single-cell libraries from cell/nucleus suspensions. | Creating cell atlases of the developing human brain; identifying novel neural subtypes [103]. |
| Targeted Enrichment Panels | (e.g., Twist Bioscience panels) Probe sets to capture and sequence specific RNA transcripts from complex samples. | Focusing sequencing resources on genes of interest (e.g., a neuroinflammatory panel) for cost-effective profiling [106]. |
The integration of classical reporter gene assays with modern sequencing technologies represents a powerful strategy for confirming gene expression and understanding its regulatory mechanisms in neuroscience. Reporter assays provide a direct, functional readout of specific regulatory elements, while RNA-seq offers an unbiased, genome-wide perspective. When combined, as in the TF-seq method, they enable researchers to seamlessly correlate upstream signaling pathway activities with downstream transcriptional outcomes. Furthermore, the advent of single-cell and multi-omic sequencing, such as SDR-seq, provides an unprecedented ability to dissect the complex relationship between genetic variation, gene regulation, and cellular function within the brain's diverse cell types. These integrated functional assays are indispensable for moving from bioinformatic predictions of disease association to mechanistic insights, ultimately accelerating the discovery of novel therapeutic targets for neurological and psychiatric disorders.
{Application Notes and Protocols}
Comparative Analysis of Cloning Techniques: Pros, Cons, and Ideal Use-Cases for Neuroscience
Within modern neuroscience research, cloning technologies have emerged as fundamental tools for dissecting the complexity of the nervous system. The application of molecular cloning and recombinant DNA technology enables scientists to probe genetic function, model neurological diseases, and develop novel therapeutic strategies. Molecular cloning, a versatile technique for isolating, amplifying, and producing recombinant DNA molecules, is a cornerstone of these efforts [107]. As large-scale initiatives like the BRAIN Initiative prioritize mapping neural circuits and understanding brain function, the role of precise genetic tools becomes increasingly critical [108]. This document provides a comparative analysis of prevailing cloning techniques, detailing their experimental protocols, advantages, limitations, and specific use-cases to guide their effective application in neuroscience.
Comparative Analysis of Cloning Techniques
The selection of an appropriate cloning technique is paramount to experimental success in neuroscience. Key methodologies offer distinct profiles of efficiency, payload capacity, and suitability for complex neural applications. The following table provides a quantitative and qualitative comparison of the most relevant techniques.
Table 1: Comparative Analysis of Cloning Techniques for Neuroscience Research
| Technique | Key Principle | Therapeutic/Research Payload | Efficiency/ Success Rate | Primary Pros | Primary Cons | Ideal Neuroscience Use-Cases |
|---|---|---|---|---|---|---|
| Somatic Cell Nuclear Transfer (SCNT) | Transfer of somatic nucleus into an enucleated egg cell [109] | Whole organisms; stem cells | Low (1-5% live birth rate in animals); high failure rates [110] | Creates genetically identical animals; source for autologous stem cells [109] | Technically challenging; ethically contentious; low efficiency [109] [110] | Genetically engineered animal models of neurodegenerative disease; therapeutic stem cell generation [109] |
| CRISPR-Based Modulation (e.g., CRISPR-TO) | Engineered Cas13 protein binds and transports RNA to subcellular locations [111] | RNA molecules for localized repair | Promoted ~50% greater neurite growth in 24h in injured neurons [111] | Unprecedented spatial precision; does not alter DNA [111] | New technology; long-term effects unknown; delivery to CNS | "Spatial RNA medicine" for ALS, spinal cord injury; localized neurite repair and outgrowth [111] |
| Recombinant Adeno-Associated Virus (rAAV) Production | Packaging of recombinant DNA into AAV capsids for gene delivery | Therapeutic genes (e.g., for gene therapy) | Varies; high interest for immune-evading capsids [76] | Established, safe profile; high transduction efficiency in neurons | Limited packaging capacity; potential immune response | Gene therapy for CNS disorders; functional gene expression in specific brain regions [76] |
| Molecular (Gene) Cloning | Isolation and amplification of specific DNA sequences in plasmids [107] | Plasmid DNA for protein expression or gene analysis | High efficiency with modern kits | Versatile, foundational technique; low-cost and standardized | Limited scale; typically used for in vitro studies | Construct generation for protein expression; promoter analysis; building blocks for complex genetic engineering [107] |
Experimental Protocols
Protocol 1: CRISPR-TO for Targeted Neuronal RNA Localization and Repair
The CRISPR-TO (CRISPR-based Targeted Orthotopic localization) system represents a novel class of spatial RNA medicine, enabling precise repair and regrowth in damaged neurons by delivering RNA to specific subcellular locations [111].
Application Note: This protocol is ideal for investigating neurite outgrowth, synaptic repair, and responses to neuronal injury in vitro. It avoids permanent genomic changes, instead modulating local RNA presence to influence cell repair mechanisms.
Materials & Reagents
- CRISPR-Cas13d Protein: Catalytically inactive or impaired version engineered for RNA binding without cleavage.
- Localization Signal Peptides: Synthesized peptides (e.g., nuclear localization signal, neurite-targeting signal) to function as molecular "zip codes".
- Target RNA Cargo: In vitro transcribed or synthetic RNA molecules of interest (e.g., growth-promoting RNAs like those identified in screening [111]).
- Primary Mouse or Human Neurons: Cultured in appropriate medium.
- Transfection Reagent: Compatible with neuronal cells for ribonucleoprotein (RNP) complex delivery.
Procedure
- Complex Formation: Pre-incubate the CRISPR-Cas13d protein with both the target RNA cargo and the selected localization signal peptide at a molar ratio of 1:2:1 (Cas13:RNA:peptide) for 30 minutes at room temperature to form the functional RNP complex.
- Neuronal Transfection: Introduce the RNP complexes into cultured primary neurons using a transfection method suitable for sensitive cells.
- Induction of Injury: If modeling injury, apply the relevant insult (e.g., oxidative stress, mechanical injury) to the culture 4-6 hours post-transfection.
- Fixation and Staining: At the desired timepoint (e.g., 24 hours post-transfection for neurite outgrowth assays [111]), fix cells and perform immunocytochemistry using antibodies against markers such as MAP2 for dendrites and Tau for axons.
- Imaging and Quantification: Acquire high-resolution images of neurons. Quantify parameters like total neurite length, number of branches, and fluorescence intensity of the localized RNA cargo at the target site (e.g., neurite tips) using automated image analysis software.
Protocol 2: SCNT for Generation of Neurological Disease Models
Somatic Cell Nuclear Transfer (SCNT) is used to create animal models that carry genetic mutations for human neurological diseases, providing a platform for studying disease mechanisms and therapeutic screening [109].
Application Note: This is a complex, resource-intensive procedure typically performed in specialized facilities. The resulting cloned animals allow for the study of genetic diseases in a whole-organism context, accounting for circuit-level and systemic effects.
Materials & Reagents
- Donor Somatic Cells: Fibroblasts from a donor animal, potentially genetically engineered to carry a disease-associated mutation.
- Oocyte Donors: Female animals to serve as a source of enucleated oocytes.
- Surrogate Females: To serve as recipients for the cloned embryos.
- Microscopes and Micromanipulators: For enucleation and nuclear transfer.
- Culture Media: For oocyte maturation, embryo culture, and activation.
Procedure
- Enucleation: Harvest mature oocytes from a donor. Using a micromanipulator, remove the metaphase II spindle and genetic material to create an enucleated oocyte.
- Nuclear Transfer: Isolate somatic cells (e.g., skin fibroblasts) from a donor animal with a specific genotype. Microinject the nucleus from one of these somatic cells into the enucleated oocyte.
- Activation: Treat the reconstructed oocyte with chemical or electrical stimuli to activate it and initiate embryonic development.
- Embryo Culture: Culture the resulting SCNT-derived embryos in vitro for a brief period to the early cleavage stage.
- Embryo Transfer: Surgically transfer the viable embryos into the uterus of a synchronously pseudopregnant surrogate female.
- Genotyping: Genotype the resulting offspring to confirm the presence of the desired transgene or mutation and to verify genetic identity with the somatic cell donor.
Diagram: SCNT Workflow for Neurological Disease Models
The Scientist's Toolkit: Research Reagent Solutions
Successful implementation of cloning protocols in neuroscience requires a suite of reliable reagents and tools. The following table details essential components for key experiments.
Table 2: Essential Research Reagents for Neuroscience Cloning Applications
| Reagent / Tool | Function | Example Use-Case in Neuroscience |
|---|---|---|
| GenScript Gene Synthesis | Custom de novo synthesis of DNA sequences with high accuracy and codon optimization [76] | Rapid construction of engineered AAV capsid variants for improved CNS targeting [76]. |
| CRISPR-Cas13d System | RNA-targeting CRISPR system that can be engineered to bind and transport RNA without cutting it [111]. | CRISPR-TO platform for localizing RNA therapeutics to sites of neuronal damage [111]. |
| Recombinant Antibodies | Highly specific affinity reagents produced via recombinant DNA technology. | Detection and validation of cell-type-specific markers (e.g., GFAP for astrocytes) in cloned brain tissue or organoids. |
| AAV Capsid Libraries | Diverse collections of AAV capsid variants for screening and selection. | In vivo screening to identify novel capsids that efficiently cross the blood-brain barrier or target specific neural cell types [76]. |
| Stem Cell Lines | Pluripotent stem cells (e.g., iPSCs) capable of differentiation into various neural lineages. | Source for generating patient-specific neurons for disease modeling or as a starting point for therapeutic cloning approaches [109]. |
The strategic application of cloning technologies is instrumental in advancing neuroscience. From creating precise animal models of Parkinson's or Huntington's disease via SCNT to delivering reparative RNA molecules with subcellular precision using CRISPR-TO, each technique offers a unique path to discovery. Future progress hinges on the continued refinement of these tools—improving the efficiency of SCNT, expanding the cargo capacity and targeting of viral vectors, and validating the therapeutic potential of spatial RNA medicine in vivo. The integration of these cloning methodologies with other cutting-edge approaches, such as single-cell sequencing and advanced imaging, will undoubtedly provide a more holistic and mechanistic understanding of the brain in health and disease.
The integration of molecular cloning and recombinant DNA technology has fundamentally transformed neuroscience research, enabling precise dissection of neural protein function in health and disease. This protocol details a comprehensive methodology for investigating S-palmitoylation, a dynamic lipid modification that regulates synaptic proteins, and validating its functional consequences using advanced electrophysiology. The reversible nature of S-palmitoylation, catalyzed by palmitoyl acyltransferases (ZDHHC enzymes) and reversed by depalmitoylases (APTs, PPTs, ABHDs), allows it to serve as a rapid regulatory mechanism for controlling protein localization, stability, and function at synapses [112]. Dysregulation of this process has been implicated in numerous neurological disorders, making it an emerging therapeutic target [112].
Molecular cloning provides the essential foundation for these investigations by enabling the construction of specific genetic tools. Techniques such as restriction enzyme cloning, Gibson Assembly, and Gateway cloning allow researchers to create plasmids expressing wild-type and mutant forms of neural proteins, tag them for detection and localization, and manipulate the enzymes that control their palmitoylation status [113] [51]. This molecular toolbox makes it possible to establish causal relationships between palmitoylation and neuronal function within the context of a broader research program focused on recombinant DNA technology in neuroscience.
The following application notes provide a structured framework for designing and executing experiments that bridge molecular manipulations with functional validation in neuronal systems. We present detailed protocols for assessing protein palmitoylation, practical methodologies for electrophysiological recording, and integrated approaches for data analysis that collectively enable a comprehensive understanding of how post-translational modifications regulate neural circuit function.
Background and Significance
S-Palmitoylation in Synaptic Function
S-palmitoylation represents a crucial regulatory mechanism in synaptic plasticity, the cellular foundation of learning and memory. Recent research has demonstrated that induction of long-term potentiation (LTP) results in protein-specific palmitoylation changes without altering global palmitoylation levels [114]. Key synaptic proteins including synaptophysin, PSD95, and neurochondrin display distinct temporal patterns of palmitoylation in response to neuronal activity, suggesting precise regulatory control over specific synaptic elements [114].
Mass spectrometry analyses of synaptoneurosomes have revealed a neuronal palmitoylome comprising over 700 proteins, with neuronal stimulation inducing predominant depalmitoylation events [114]. These dynamically modified proteins are functionally associated with synaptic vesicle cycling, cytoskeletal dynamics, and neurotransmitter release, positioning palmitoylation as a master regulator of synaptic transmission machinery. Interestingly, synaptoneurosomes contain active palmitoylation machinery capable of supporting rapid, target-specific responses to NMDA receptor activation, highlighting the spatial and temporal precision of this modification system [114].
Molecular Tools for Neuroscience Research
Molecular cloning technologies provide the foundation for manipulating and studying palmitoylation processes. Restriction enzyme cloning utilizing enzymes such as EcoRI and HindIII enables precise DNA fragment isolation and insertion into plasmid vectors [113] [51]. More advanced techniques like Gibson Assembly and Golden Gate cloning allow seamless assembly of multiple DNA fragments, greatly facilitating the construction of complex genetic tools [51].
These approaches enable creation of plasmids for expressing wild-type and cysteine mutant proteins that cannot be palmitoylated, tagged versions of proteins for localization and detection, and manipulation of ZDHHC enzymes and depalmitoylases that control the palmitoylation cycle. When combined with bacterial transformation and selection systems (e.g., blue-white screening), researchers can generate the specific molecular tools needed to interrogate how palmitoylation regulates neural protein function [113] [51].
Application Notes
Protein-Specific Palmitoylation Dynamics During Synaptic Plasticity
Investigation of palmitoylation dynamics during synaptic plasticity requires specialized methodologies capable of capturing rapid, protein-specific changes. The acyl-biotin exchange (ABE) assay provides a robust biochemical approach for quantifying palmitoylation levels of specific proteins under different stimulation conditions [114]. This method involves replacing palmitate groups with biotin, followed by affinity purification and immunoblotting for proteins of interest.
Induction of chemical long-term potentiation (cLTP) in neuronal cultures using picrotoxin, forskolin, and rolipram generates a tetanic-like stimulation that triggers protein-specific palmitoylation changes without affecting global palmitoylation levels [114]. This approach has revealed that synaptophysin and PSD95 display distinct temporal patterns of palmitoylation following LTP induction, suggesting differential regulatory mechanisms for presynaptic and postsynaptic proteins.
Experimental models ranging from primary neuronal cultures to hippocampal slice preparations and synaptoneurosomes provide complementary information about palmitoylation dynamics at different biological scales [114]. Mass spectrometry of synaptoneurosomes has proven particularly valuable for identifying novel palmitoylation targets and mapping stimulus-dependent changes across the neuronal palmitoylome.
Table 1: Key Proteins with Activity-Dependent Palmitoylation Changes
| Protein | Synaptic Location | Palmitoylation Response | Functional Role |
|---|---|---|---|
| Synaptophysin | Presynaptic | Increased after cLTP | Synaptic vesicle cycling |
| PSD95 | Postsynaptic | Temporal pattern after LTP | Scaffolding protein |
| Neurochondrin | Postsynaptic | Altered after cLTP | LTP maintenance |
| VAMP2 | Presynaptic | Stimulation-dependent | Vesicle fusion |
| GluR1 | Postsynaptic | Activity-regulated | AMPA receptor trafficking |
Electrophysiological Validation Using High-Density Microelectrode Arrays
High-density microelectrode arrays (HD-MEAs) represent a transformative technology for functional validation of palmitoylation effects on neuronal activity. Modern CMOS-based HD-MEA systems offer unprecedented spatial and temporal resolution, with some devices featuring >3000 electrodes per mm² and simultaneous readout of over 30,000 channels at 70 kHz sampling rates [115]. This technological advancement enables researchers to monitor neuronal activity across multiple scales - from subcellular compartments to entire networks - over extended time periods.
HD-MEA applications in palmitoylation research include assessing basal excitatory and inhibitory synaptic transmission, LTP induction and maintenance, and network-level synchrony following manipulation of palmitoylation pathways [114] [115]. The capability for both recording and stimulation within the same platform provides a powerful tool for probing causal relationships between palmitoylation status and functional synaptic plasticity.
Complementary approaches using human induced pluripotent stem cell (hiPSC)-derived neurons on multielectrode array (MEA) platforms enable investigation of human-specific aspects of neuronal function and pharmacological responses [116]. These systems have demonstrated sensitivity to neuromodulators such as opioid receptor agonists, establishing their utility for studying regulated neuronal signaling pathways [116].
Table 2: HD-MEA Specifications for Synaptic Function Analysis
| Parameter | Low-Density MEA | High-Density MEA | Application in Palmitoylation Studies |
|---|---|---|---|
| Electrode density | 10-100/mm² | >3000/mm² | Subcellular resolution of AP propagation |
| Simultaneous channels | 60-256 | Up to 33,840 | Large-scale network activity mapping |
| Electrode size | 10-30 μm | ~11 μm | Single-unit isolation |
| Spatial resolution | Cellular | Subcellular | Dendritic vs. axonal signaling |
| Throughput | Medium | High | Multiple treatment conditions |
Protocols
Acyl-Biotin Exchange (ABE) Assay for Protein Palmitoylation
Principle and Applications
The ABE assay allows biochemical quantification of palmitoylation levels for specific proteins of interest. This method exploits the covalent nature of the thioester bond linking palmitate to cysteine residues, replacing the palmitoyl groups with biotin for sensitive detection and purification [114]. The protocol can be applied to various sample types, including neuronal cultures, brain homogenates, and synaptoneurosomes.
Reagents and Solutions
- Lysis Buffer: 150 mM NaCl, 50 mM Tris-HCl (pH 7.4), 5 mM EDTA, 1% Triton X-100, supplemented with protease inhibitors
- Blocking Buffer: 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 2.5% SDS, 10 mM N-ethylmaleimide (NEM)
- Hydroxylamine Solution: 1 M hydroxylamine (pH 7.4), 1 mM HPDP-biotin, 0.2% Triton X-100
- Control Solution: 50 mM Tris (pH 7.4) instead of hydroxylamine
- NeutrAvidin Agarose beads for affinity capture
Step-by-Step Procedure
- Sample Preparation: Lyse cells or tissue in ice-cold lysis buffer. Clear lysates by centrifugation at 16,000 × g for 15 minutes at 4°C.
- Free Thiol Blocking: Incubate lysates with blocking buffer for 2-4 hours at 4°C with gentle rotation. Remove excess NEM by acetone precipitation or desalting columns.
- Palmitate Cleavage and Biotin Labeling: Split samples into two equal aliquots. Treat one with hydroxylamine solution (test condition) and the other with control solution (negative control). Incubate for 1-2 hours at room temperature.
- Biotin Capture: Incubate samples with NeutrAvidin agarose beads overnight at 4°C with rotation.
- Washing and Elution: Wash beads extensively with lysis buffer. Elute bound proteins with 2× SDS-PAGE sample buffer containing β-mercaptoethanol.
- Detection: Analyze eluates by immunoblotting for proteins of interest. Compare hydroxylamine-treated and control samples to quantify palmitoylation levels.
Critical Considerations
- Include appropriate controls (no hydroxylamine, no biotin) to account for non-specific binding.
- Optimize NEM concentration and blocking time to ensure complete thiol blocking without excessive protein modification.
- Normalize results to total protein levels in input samples for accurate quantification.
- For temporal studies, process samples rapidly and include protease and deacylase inhibitors to preserve native palmitoylation states.
Functional Assessment Using High-Density Microelectrode Arrays
HD-MEA Experimental Setup
Modern HD-MEA systems integrate recording electrodes, amplification circuits, and analog-to-digital converters on a single chip, enabling high-signal-to-noise ratio recordings [115]. Platform selection should consider specific experimental needs, including spatial resolution, channel count, and multimodal integration capabilities.
Neuronal Culture on HD-MEAs
- Surface Preparation: Treat MEA surfaces with poly-D-lysine (0.1 mg/mL) and laminin (2 μg/mL) to promote neuronal adhesion.
- Cell Plating: Plate primary rodent neurons or hiPSC-derived neurons at densities of 800-1200 cells/mm², optimized for network formation while maintaining single-cell resolution.
- Maintenance: Culture neurons in neurobasal medium with appropriate supplements, maintaining cultures at 35-37°C with 5% CO₂.
- Maturation: Allow 14-28 days in vitro (DIV) for synaptic maturation before experiments, with regular medium changes.
Recording and Stimulation Parameters
- Data Acquisition: Sample at ≥20 kHz per channel to adequately capture action potential waveforms.
- Signal Filtering: Apply bandpass filtering (300-3000 Hz) for spike detection and wider bandpass (1-5000 Hz) for local field potentials.
- Electrical Stimulation: Use bipolar current pulses (10-100 μA, 100-500 μs duration) through selected electrodes for synaptic stimulation.
- LTP Induction: Apply high-frequency stimulation (100 Hz, 1s) or theta-burst stimulation (5 Hz, 10 bursts of 4 pulses at 100 Hz) to induce plasticity.
Pharmacological Manipulation of Palmitoylation
- Palmitoylation Inhibition: Apply 2-bromopalmitate (2-BP; 10-100 μM) or specific ZDHHC inhibitors to block palmitoylation.
- Depalmitoylation Inhibition: Use palmostatin B (5-20 μM) to inhibit depalmitoylases.
- Acute Modulation: Treat cultures during HD-MEA recording to assess real-time functional effects.
- Chronic Manipulation: Pre-treat cultures for 6-24 hours before recording to assess cumulative effects.
HD-MEA Functional Analysis: HD-MEA technology enables multiscale analysis of neuronal function from subcellular to network levels.
The Scientist's Toolkit
Research Reagent Solutions
Table 3: Essential Reagents for Palmitoylation-Electrophysiology Studies
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Molecular Cloning Tools | Restriction enzymes (EcoRI, HindIII), T4 DNA Ligase, TOPO vectors | Plasmid construction for protein expression and gene manipulation |
| Palmitoylation Modulators | 2-Bromopalmitate (2-BP), Palmostatin B, N-(tert-Butyl)hydroxylamine (NtBuHA) | Pharmacological manipulation of palmitoylation cycles |
| Neuronal Culture Components | Poly-D-lysine, Laminin, Neurobasal medium, B-27 supplement | Maintenance of primary neurons and hiPSC-derived neurons on MEAs |
| Detection Reagents | HPDP-biotin, NeutrAvidin agarose, N-ethylmaleimide (NEM) | Biochemical assessment of palmitoylation (ABE assay) |
| Electrophysiology Solutions | Artificial cerebrospinal fluid (ACSF), Tetrodotoxin (TTX), CNQX, AP5 | Maintenance of neuronal activity and pharmacological isolation |
Equipment and Software
Table 4: Essential Equipment for Integrated Palmitoylation-Electrophysiology Studies
| Equipment Category | Specific Examples | Key Features |
|---|---|---|
| High-Density MEA Systems | CMOS-based HD-MEA platforms, Multiwell MEA systems | High spatial resolution, simultaneous recording/stimulation, low noise |
| Molecular Biology Equipment | Thermocyclers, Electroporators, Gel electrophoresis systems | Plasmid construction, validation, and preparation |
| Imaging Systems | Confocal microscopes, Live-cell imaging systems | Validation of protein localization and trafficking |
| Data Analysis Software | Custom MATLAB scripts, Python packages (e.g., SpyKING CIRCUS) | Spike sorting, network analysis, statistical comparison |
| Cell Culture Equipment | Biosafety cabinets, CO₂ incubators, Inverted microscopes | Maintenance of neuronal cultures under sterile conditions |
Data Analysis and Integration
Electrophysiological Data Processing
Analysis of HD-MEA data requires specialized computational approaches to extract meaningful information from large-scale recordings. Spike sorting algorithms identify and classify action potentials from individual neurons, enabling tracking of single-unit activity across large networks [115]. For synaptic plasticity experiments, burst detection algorithms identify periods of high-frequency activity, while cross-correlation analysis quantifies functional connectivity between neurons.
Parameters for assessing palmitoylation-dependent effects include:
- Mean firing rates: Changes in overall network excitability
- Burst characteristics: Duration, inter-burst intervals, spikes per burst
- Synchrony metrics: Correlation coefficients between neuronal pairs
- LTP/LTD magnitude: Persistent changes in synaptic strength following induction protocols
Statistical Considerations and Experimental Design
Appropriate experimental design is crucial for reliable interpretation of palmitoylation-electrophysiology studies. Include biological replicates (different culture preparations) and technical replicates (multiple wells/MEAs per condition) to account for variability. For pharmacological experiments, include vehicle controls and concentration-response relationships to establish specificity.
Statistical approaches should account for the multivariate nature of electrophysiological data. Multivariate ANOVA can assess overall treatment effects across multiple parameters, while post-hoc tests identify specific differences. For time-series data, repeated measures ANOVA or mixed-effects models appropriately account for correlated measurements.
Troubleshooting Guide
Common Challenges and Solutions
Table 5: Troubleshooting Common Issues in Palmitoylation-Electrophysiology Studies
| Problem | Potential Causes | Solutions |
|---|---|---|
| Low signal in ABE assay | Incomplete blocking or biotinylation | Optimize NEM concentration; verify hydroxylamine activity; fresh HPDP-biotin |
| High background in ABE | Non-specific binding to beads | Increase stringency of washes; include no-hydroxylamine controls |
| Poor neuronal viability on MEAs | Suboptimal surface treatment | Test different coating protocols; verify medium components |
| Low spontaneous activity | Immature networks or poor health | Extend culture time; verify glial support; check medium components |
| Excessive network bursting | Overly dense cultures or imbalance in excitation/inhibition | Optimize plating density; adjust E/I balance pharmacologically |
| Inconsistent LTP induction | Variability in stimulation parameters | Systematically optimize stimulation location and parameters |
Palmitoylation Regulation Pathway: Dynamic palmitoylation cycles controlled by ZDHHC enzymes and depalmitoylases regulate synaptic protein localization and function, which can be quantified using HD-MEA systems.
The integrated methodology presented here - combining molecular manipulation of palmitoylation with functional validation using advanced electrophysiology - provides a powerful framework for investigating how post-translational modifications regulate neural protein function. As molecular cloning techniques continue to evolve, enabling more precise genetic manipulations, and HD-MEA technology advances toward even higher densities and multimodal integration, researchers will gain unprecedented insight into the molecular mechanisms underlying synaptic plasticity and neuronal circuit function.
The protocols and application notes detailed in this document establish a foundation for comprehensive investigation of protein palmitoylation in neuronal systems. By following these methodologies and adapting them to specific research questions, neuroscientists can systematically elucidate how reversible lipid modifications contribute to the dynamic regulation of synaptic strength, network activity, and ultimately, complex cognitive processes.
The transition from in vitro model systems to robust pre-clinical validation is a critical juncture in neuroscience drug development. Molecular cloning and recombinant DNA technologies are the bedrock of this process, enabling the precise genetic manipulation required to create physiologically relevant human-based models and therapeutic agents. This Application Note provides a detailed framework for assessing the therapeutic potential of novel interventions, from initial in vitro screening in complex models to definitive pre-clinical studies, all within the context of modern molecular biology techniques.
Quantitative Data Analysis from In Vitro Models
Quantitative data analysis is essential for examining numerical data to uncover patterns, test hypotheses, and support decision-making in drug development [117]. When comparing quantitative data between different experimental groups, the data should be summarized for each group, and the difference between means or medians must be computed [118].
Table 1: Summary of Quantitative Data from Gorilla Chest-Beating Study [118] This table exemplifies the standard format for presenting descriptive statistics when comparing two groups.
| Group (beats per 10 h) | Mean | Standard Deviation | Sample Size (n) |
|---|---|---|---|
| Younger Gorillas | 2.22 | 1.270 | 14 |
| Older Gorillas | 0.91 | 1.131 | 11 |
| Difference | 1.31 | --- | --- |
Table 2: In Vitro Efficacy Data for Anti-Lyn siRNA in Glioma Stem-like Cells [119] This table summarizes hypothetical quantitative data for a key experiment validating a therapeutic target.
| Cell Model | Lyn Expression Status | Treatment | Mean Cell Viability (%) ± SD | n | p-value (vs. Untreated Control) |
|---|---|---|---|---|---|
| Glioma Stem-like Cells | Lyn-positive | Untreated Control | 100 ± 8.5 | 9 | --- |
| Anti-Lyn siRNA | 62.5 ± 7.1 | 9 | < 0.001 | ||
| Control Cell Line | Lyn-negative | Untreated Control | 100 ± 6.3 | 6 | --- |
| Anti-Lyn siRNA | 98.2 ± 5.9 | 6 | 0.45 |
Visualization of Comparative Data: For comparative quantitative data, boxplots are an excellent choice as they summarize the distribution using the median, quartiles, and potential outliers, allowing for easy visual comparison between groups [118]. Dot charts are also effective for displaying individual data points across groups, especially when jittering is used to prevent overplotting [118].
Experimental Protocols
Objective: To reprogram patient somatic cells into induced Pluripotent Stem Cells (iPSCs) for the creation of complex in vitro models (CIVMs) of rare neurological diseases.
Materials:
- Source Tissue: Patient dermal fibroblasts or peripheral blood mononuclear cells (PBMCs).
- Reprogramming Factors: Sendai viral vectors (non-integrating) encoding OCT4, SOX2, KLF4, and c-MYC.
- Culture Medium: Pluripotent stem cell maintenance medium (e.g., mTeSR or equivalent).
- Matrix: Growth factor-reduced Matrigel or recombinant Laminin-521.
- Equipment: Class II biological safety cabinet, humidified CO2 incubator, water bath, centrifuge, inverted microscope.
Methodology:
- Cell Preparation: Isolate and expand patient fibroblasts or PBMCs in their respective culture media. Passage cells as needed until a sufficient number are obtained.
- Viral Transduction: On day 0, plate 5 x 10^4 to 1 x 10^5 target cells in a 6-well plate. On day 1, transduce cells with the Sendai virus vectors at an appropriate Multiplicity of Infection (MOI) in the presence of polybrene (5-8 µg/mL) to enhance transduction efficiency.
- Media Change: After 24 hours, replace the transduction medium with fresh cell-specific growth medium.
- Cell Passaging: On day 7 post-transduction, harvest the transduced cells using enzymatic dissociation (e.g., TrypLE). Re-plate the cells onto a Matrigel-coated 6-well plate at a high density in fibroblast or PBMC medium.
- Medium Switch: 24 hours after passaging, carefully switch the medium to pre-warmed pluripotent stem cell maintenance medium. Continue feeding the cells daily with this medium.
- Colony Picking: After 3-4 weeks, distinct iPSC colonies will become visible. Manually pick and transfer individual, morphologically undifferentiated colonies onto new Matrigel-coated plates for expansion.
- Validation: Characterize established iPSC lines via immunocytochemistry (for NANOG, OCT4, SSEA4), flow cytometry, and/or qRT-PCR to confirm pluripotency marker expression. Perform karyotyping to ensure genomic integrity.
Objective: To evaluate the efficacy of a dendrimer-based siRNA delivery system targeting Lyn kinase in patient-derived glioma stem-like cells (GSCs) cultured as 3D tumor spheroids.
Materials:
- Cells: Patient-derived glioma stem-like cells (GSCs).
- Therapeutic Agent: Phosphorus dendrimer-based nanoformulation complexed with anti-Lyn siRNA or scrambled siRNA control.
- Culture Medium: Serum-free neural stem cell medium supplemented with EGF and bFGF.
- 3D Culture Plates: Low-adherence, U-bottom 96-well plates for spheroid formation.
- Assay Kits: Cell Titer-Glo 3D Viability Assay kit.
- Equipment: Luminometer, microplate shaker, centrifuge.
Methodology:
- Spheroid Generation: Harvest GSCs as a single-cell suspension. Seed 2 x 10^3 cells per well in 100 µL of complete medium into a low-adherence 96-well plate. Centrifuge the plate at low speed (e.g., 300 x g for 3 minutes) to aggregate cells at the bottom of the wells.
- Spheroid Culture: Incubate the plate for 72-96 hours to allow for the formation of compact, single spheroids per well.
- Treatment Formulation: Complex the dendrimer with anti-Lyn siRNA or scrambled siRNA at the optimal N/P (nitrogen-to-phosphate) ratio in serum-free medium. Incubate for 20 minutes at room temperature to allow for polyelectrolyte complex formation.
- Therapeutic Intervention: After spheroid formation, carefully add 100 µL of the dendrimer-siRNA complexes (or dendrimer-only and untreated controls) to the respective wells. Gently swirl the plate to mix.
- Incubation: Incubate the treated spheroids for 72-120 hours.
- Viability Assessment: Equilibrate the Cell Titer-Glo 3D reagent to room temperature. Add 100 µL of the reagent to each well. Place the plate on an orbital shaker for 5 minutes to induce cell lysis. Incubate the plate for 25 minutes at room temperature to stabilize the luminescent signal.
- Quantification: Record the luminescence using a luminometer. Normalize the data to the untreated control (100% viability) and calculate the percentage cell viability for each treatment group. Perform statistical analysis using an unpaired t-test or ANOVA.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Molecular Cloning and Cell-Based Therapeutic Assessment
| Item | Function & Application |
|---|---|
| Recombinant DNA Vectors (Plasmids) | Engineered DNA molecules used as vehicles to artificially carry foreign genetic material into a host cell for cloning (replication) or expression of a gene of interest [20]. |
| Restriction Endonucleases & DNA Ligase | Molecular scissors and glue. Restriction enzymes cut DNA at specific sequences, while DNA ligase joins DNA fragments together, forming recombinant DNA molecules [20]. |
| Polymerase Chain Reaction (PCR) Reagents | Enzymes (e.g., Taq polymerase) and nucleotides to amplify specific DNA sequences exponentially from a small initial sample for cloning, analysis, or detection [120]. |
| Dendrimer-Based Nanoformulations | Synthetic, highly branched polymers used as nanocarriers for the delivery of nucleic acid therapeutics (siRNA, DNA) into cells, offering high transfection efficiency and potential for functionalization [119]. |
| Induced Pluripotent Stem Cells (iPSCs) | Patient-derived somatic cells that have been reprogrammed into an embryonic-like pluripotent state. They enable the generation of patient-specific disease models, including brain organoids, for personalized therapeutic screening [121]. |
| 3D Extracellular Matrix (ECM) Hydrogels | Biomimetic scaffolds (e.g., Matrigel, collagen) that provide a three-dimensional environment for culturing cells as spheroids or organoids, promoting more in vivo-like cell behavior and signaling [119] [121]. |
| Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas9 Systems | A revolutionary recombinant DNA technology for precise genome editing. It allows for the knockout, knock-in, or correction of disease-associated mutations in cell lines and model organisms [20]. |
Conclusion
Molecular cloning and recombinant DNA technology have irrevocably transformed neuroscience, providing the tools to dissect the molecular underpinnings of brain function and disease with unparalleled precision. From enabling optogenetics to probe neural circuits to facilitating the study of neurodegenerative disease mechanisms and creating novel therapeutic candidates like recombinant antibodies, these techniques are pillars of modern neurobiological research. The future points toward even greater integration with emerging technologies such as CRISPR-Cas9 for precision gene editing, advanced synthetic biology for constructing complex neural pathways, and automated high-throughput systems. This progression will undoubtedly accelerate the development of next-generation treatments for neurological and psychiatric disorders, solidifying the central role of genetic engineering in unlocking the brain's mysteries and improving human health.
References
- 1. Brief guide to gene cloning - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12302143/]
- 2. Molecular Cloning Technology - Past, Present and Future [https://www.neb.com/en-us/tools-and-resources/feature-articles/foundations-of-molecular-cloning-past-present-and-future?srsltid=AfmBOoqQLck-0JvQBqtsLpEeXim2k7P-btgGhF-K0YpcRmHZjTLHF4gZ]
- 3. A comparative review of DNA assembly strategies [https://www.sciencedirect.com/science/article/pii/S2452014425002493]
- 4. BRAIN 2025: A Scientific Vision - BRAIN Initiative - NIH [http://braininitiative.nih.gov/vision/nih-brain-initiative-reports/brain-2025-scientific-vision]
- 5. Comparison of Analytic Methods for Quantitative Real-Time ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4642823/]
- 6. The Influence of Restriction Endonucleases for Cloning [https://auctoresonline.org/article/the-influence-of-restriction-endonucleases-for-cloning]
- 7. Restriction Enzyme Cloning [https://www.snapgene.com/guides/restriction-enzyme-cloning]
- 8. Advances in Diversity, Evolutionary Dynamics and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12114051/]
- 9. AAV vector development, back to the future [https://www.sciencedirect.com/science/article/pii/S1525001625002734]
- 10. Spatial genomics of AAV vectors reveals mechanism ... [https://www.nature.com/articles/s41587-025-02565-4]
- 11. Traditional Cloning Quick Guide [https://www.neb.com/en-us/tools-and-resources/usage-guidelines/cloning-guide?srsltid=AfmBOoomt7TS8soHfkvDeeEIJfj3w_HKjHS9ZS45C7hFcvn2-880l3Fi]
- 12. Molecular Cloning Technology - Past, Present and Future [https://www.neb.com/en-us/tools-and-resources/feature-articles/foundations-of-molecular-cloning-past-present-and-future?srsltid=AfmBOopEpPwNTYWV3Q6zy_bXI6T0zSfBF8lAfy-ECZutupVuMhWRQW3Z]
- 13. History of insulin - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3714061/]
- 14. Recombinant Drugs | National Museum of American History [https://americanhistory.si.edu/collections/object-groups/birth-of-biotech/recombinant-drugs]
- 15. Recombinant DNA strategies in genetic neurological diseases [https://pubmed.ncbi.nlm.nih.gov/6310392/]
- 16. Scientists design gene delivery systems for cells in the ... [https://www.nih.gov/news-events/news-releases/scientists-design-gene-delivery-systems-cells-brain-spinal-cord]
- 17. 100 Years of Insulin [https://www.fda.gov/about-fda/fda-history-exhibits/100-years-insulin]
- 18. Human Insulin: History, Recent Advances, and Expression ... [https://bmrat.org/index.php/BMRAT/article/view/692]
- 19. New Research on AI as a Diagnostic Tool to Be Featured ... [https://www.newswise.com/articles/new-research-on-ai-as-a-diagnostic-tool-to-be-featured-at-amp-2025]
- 20. Role of Recombinant DNA Technology to Improve Life - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5178364/]
- 21. Introducing cloned genes into cultured neurons providing novel in vitro models for neuropathology and neurotoxicity studies - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7055714/]
- 22. Unleashing the Power of Recombinant DNA Technology: Gene Cloning, Expression and Engineering - Life in the Lab [https://www.thermofisher.com/blog/life-in-the-lab/unleashing-the-power-of-recombinant-dna-technology-gene-cloning-expression-and-engineering/]
- 23. Recombinant DNA - Wikipedia [https://en.wikipedia.org/wiki/Recombinant_DNA]
- 24. Spatial transcriptomics in neuroscience [https://www.nature.com/articles/s12276-023-01093-y]
- 25. PCR Cloning Method [https://www.neb.com/en-us/applications/cloning-and-synthetic-biology/pcr-cloning?srsltid=AfmBOooBqJ5pPO31LwV4ya-OxXPAp8D1O6X2tXMY2Q9otoZsVv56Fr_m]
- 26. Plasmid Cloning by PCR (with Protocols) [https://www.addgene.org/protocols/pcr-cloning/]
- 27. Advanced Approaches: PCR, Traditional Cloning, and ... [https://blog.edvotek.com/2025/08/06/advanced-approaches-pcr-traditional-cloning-and-golden-gate-assembly-compared/]
- 28. Molecular cloning using polymerase chain reaction, an ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4350901/]
- 29. Revolutionizing Molecular cloning [https://pmc.ncbi.nlm.nih.gov/articles/PMC11906075/]
- 30. A PCR-independent, annealing-free cloning method for the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11659769/]
- 31. Plasmid Cloning by Restriction Enzyme Digest ... [https://www.addgene.org/protocols/subcloning/]
- 32. The Application of the Gibson Assembly Method in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10301892/]
- 33. Gibson Assembly vs Golden Gate: A Complete Guide to ... [https://www.clyte.tech/post/gibson-assembly-vs-golden-gate-a-complete-guide-to-choosing-the-right-cloning-method?srsltid=AfmBOoq3KGdPwewR-6Jw_6KGrJsldeWF2kI8F3RW6p47xvQoFqnfjk-C]
- 34. Gateway Cloning Protocols [https://www.thermofisher.com/us/en/home/life-science/cloning/gateway-cloning/protocols.html]
- 35. Gateway Recombinational Cloning - PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC5935001/]
- 36. Golden Gate Assembly vs. Gibson Assembly: DNA Cloning ... [https://synapse.patsnap.com/article/golden-gate-assembly-vs-gibson-assembly-dna-cloning-methods-compared]
- 37. Codon Optimization for Increased Protein Expression [https://www.genscript.com/codon-optimization-for-increased-protein-expression.html]
- 38. Plasmids 101: Codon usage bias [https://blog.addgene.org/plasmids-101-codon-usage-bias]
- 39. Link Between Individual Codon Frequencies and Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11546221/]
- 40. Codon optimization with deep learning to enhance protein ... [https://www.nature.com/articles/s41598-020-74091-z]
- 41. Comparative Analysis of Codon Optimization Tools [https://pmc.ncbi.nlm.nih.gov/articles/PMC12010093/]
- 42. Codon Optimization Tool [https://en.vectorbuilder.com/tool/codon-optimization.html]
- 43. Codon Table [https://en.vectorbuilder.com/tool/codon-table.html]
- 44. Codon Usage Frequency Table(chart) [https://www.genscript.com/tools/codon-frequency-table]
- 45. Codon Optimization Tool | IDT [https://www.idtdna.com/pages/tools/codon-optimization-tool]
- 46. Independent Optical Excitation of Distinct Neural Populations [https://pmc.ncbi.nlm.nih.gov/articles/PMC3943671/]
- 47. Optogenetics Guide [https://www.addgene.org/guides/optogenetics/]
- 48. Temporal Resolution of ChR2 and Chronos in an Optogenetic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4465525/]
- 49. Crystal structure of the red light-activated ... [https://www.nature.com/articles/s41467-018-06421-9]
- 50. Classical Recombinant DNA Cloning [https://pubmed.ncbi.nlm.nih.gov/36853452/]
- 51. Molecular cloning: Techniques and applications [https://www.abcam.com/en-us/knowledge-center/dna-and-rna/molecular-cloning-techniques-and-applications]
- 52. HulaChrimson: A Chrimson-like cation channelrhodopsin ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12399511/]
- 53. Optogenetic tools and their applications for therapeutic ... [https://www.sciencedirect.com/science/article/pii/S0098299725000524]
- 54. Notices of Funding Opportunities - BRAIN Initiative - NIH [http://braininitiative.nih.gov/funding/funding-opportunities]
- 55. Molecular cloning, expression, and functional analyses of ... [https://pubmed.ncbi.nlm.nih.gov/39800193/]
- 56. Molecular cloning, expression, and functional analyses of ... [https://www.sciencedirect.com/science/article/abs/pii/S0378111925000307]
- 57. Glioblastoma at the crossroads: current understanding and ... [https://www.nature.com/articles/s41392-025-02299-4]
- 58. Scientists wipe out aggressive brain cancer tumors by ... [https://ufhealth.org/news/2025/scientists-wipe-out-aggressive-brain-cancer-tumors-by-targeting-cellular-motors]
- 59. Psychedelics Reverse the Polarity of Long-Term Synaptic ... [https://www.eneuro.org/content/12/10/ENEURO.0047-25.2025]
- 60. All-optical voltage interrogation for probing synaptic ... [https://www.nature.com/articles/s41467-025-63867-4]
- 61. Computational protocol for modeling and analyzing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11915160/]
- 62. Blue-shifted ancyromonad channelrhodopsins for multiplex ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12448745/]
- 63. Evolution of Light-Sensitive Proteins in Optogenetic ... [https://www.mdpi.com/2227-9059/13/2/429]
- 64. Channelrhodopsin fluorescent tag replacement for clinical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10111946/]
- 65. An engineered channelrhodopsin optimized for axon ... [https://www.nature.com/articles/s42003-021-01977-7]
- 66. Optogenetics: A Novel Therapeutic Avenue for Age-Related ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12467667/]
- 67. Optogenetic induction of subcellular Ca2+ events in ... [https://www.nature.com/articles/s42003-025-08924-w]
- 68. Overexpression of enhanced yellow fluorescent protein fused ... [https://pubmed.ncbi.nlm.nih.gov/39584396/]
- 69. Blue-shifted ancyromonad channelrhodopsins for multiplex ... [https://elifesciences.org/articles/106508]
- 70. The Plasmid Cloning Cycle [https://www.snapgene.com/guides/the-plasmid-cloning-cycle]
- 71. Molecular Cloning: A Detailed Introduction [https://www.goldbio.com/blogs/articles/molecular-cloning-detailed-intro?srsltid=AfmBOoo4wIwrUj2YrIkgcjxGy5gZijV1150K-2Cm2C4lTwFzPkwW_2E6]
- 72. Traditional Cloning Basics | Thermo Fisher Scientific - ES [https://www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/molecular-cloning/cloning/traditional-cloning-basics.html]
- 73. Recombinant DNA - The Cell - NCBI Bookshelf - NIH [https://www.ncbi.nlm.nih.gov/books/NBK9950/]
- 74. Molecular Cloning Techniques [https://www.addgene.org/mol-bio-reference/cloning/]
- 75. Recombinant DNA technology – Steps, Methods & Examples [https://www.evitria.com/journal/recombinant-antibodies/recombinant-dna-technology/]
- 76. GenScript Life Science Research Grant Program 2025 [https://www.genscript.com/grantprogram-2025.html]
- 77. Efficient Generation of hiPSC Neural Lineage Specific ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4542987/]
- 78. Designing highly multiplex PCR primer sets with Simulated ... [https://www.nature.com/articles/s41467-022-29500-4]
- 79. DeGenPrime provides robust primer design and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11001487/]
- 80. Effective primer design for genotype and subtype detection of ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-025-06251-9]
- 81. Restriction Site - an overview [https://www.sciencedirect.com/topics/neuroscience/restriction-site]
- 82. Molecular Cloning Guide [https://www.promega.com/resources/guides/nucleic-acid-analysis/subcloning/]
- 83. Troubleshooting Guide for Cloning [https://www.neb.com/en-us/tools-and-resources/troubleshooting-guides/troubleshooting-guide-for-cloning?srsltid=AfmBOoo8Kaaq-US_BKUWtcsiKKFKeXgLtf2JISNEInz6H47BwNcS2rfQ]
- 84. Cloning Troubleshooting Guide [https://www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/molecular-cloning/cloning/cloning-troubleshooting-guide.html]
- 85. Optimize Your DNA Ligation with 7 Must-Have Tips [https://www.thermofisher.com/us/en/home/brands/thermo-scientific/molecular-biology/molecular-biology-learning-center/molecular-biology-resource-library/spotlight-articles/optimize-dna-ligase.html]
- 86. 11 Reasons Why Your Plasmid Yield is Low & Easy Fixes [https://bitesizebio.com/13514/boost-plasmid-yield/]
- 87. Simple Tips to Improve your Cloning Efficiency [https://www.neb.com/en-us/tools-and-resources/usage-guidelines/getting-started-with-molecular-cloning-simple-guidelines-to-improve-your-cloning-efficiency?srsltid=AfmBOoqpaSUzP7M-Gb4vcqxH5e_WirQKsszSjFQjofk7gcXlu-t5OWLW]
- 88. Recombinant Gene - an overview [https://www.sciencedirect.com/topics/neuroscience/recombinant-gene]
- 89. CRISPR-StAR enables high-resolution genetic screening ... [https://www.nature.com/articles/s41587-024-02512-9]
- 90. Automated Plasmid Purification [https://www.thermofisher.com/us/en/home/life-science/dna-rna-purification-analysis/plasmid-isolation/automated-plasmid-purification.html]
- 91. Advancing Plasmid DNA Purification at Scale - The Scientist [https://www.the-scientist.com/advancing-plasmid-dna-purification-at-scale-73304]
- 92. Top 10 Industry Predictions for 2025 [https://www.cryoport.com/bio-blog-top-10-industry-predictions-for-2025/]
- 93. IMCS at SLAS 2025 [https://imcstips.com/slas2025/]
- 94. Automation Protocol for Plasmid DNA Extraction from E. coli [https://www.protocols.io/view/automation-protocol-for-plasmid-dna-extraction-fro-d49y8z7w.html]
- 95. Molecular Cloning Market to Attain USD 8.89 Billion by [https://www.globenewswire.com/news-release/2025/11/12/3186444/0/en/Molecular-Cloning-Market-to-Attain-USD-8-89-Billion-by-2034-Driven-by-Expanding-Healthcare-Applications-and-Innovation.html]
- 96. A Next-Generation Sequencing Primer—How Does It Work ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5944141/]
- 97. New study challenges gold standard for validating DNA ... [https://www.genome.gov/news/news-release/New-study-challenges-gold-standard-for-validating-DNA-sequencing-results]
- 98. A Molecular Cloning and Sanger Sequencing-based ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9090521/]
- 99. A practical guide to sequencing in neuropsychiatric research [https://www.nature.com/articles/s44277-025-00041-0]
- 100. Simultaneous Pathway Activity Inference and Gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5032147/]
- 101. Bioluminescent Reporters | Reporter Gene Applications [https://www.promega.com/resources/guides/cell-biology/bioluminescent-reporters/]
- 102. Gene reporter assays [https://www.bmglabtech.com/en/blog/gene-reporter-assays/]
- 103. Applications of single-cell RNA sequencing in drug discovery ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10141847/]
- 104. Functional phenotyping of genomic variants using joint ... [https://www.nature.com/articles/s41592-025-02805-0]
- 105. Cloning Gene Variants and Reporter Assays [https://pubmed.ncbi.nlm.nih.gov/26498618/]
- 106. Trending Applications in RNA Sequencing [https://www.twistbioscience.com/blog/science/trending-rna-seq-applications]
- 107. MiniResource Brief guide to gene cloning [https://www.sciencedirect.com/science/article/pii/S1016847825000585]
- 108. The BRAIN Initiative: developing technology to catalyse ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4387507/]
- 109. Why Clone? [https://learn.genetics.utah.edu/content/cloning/whyclone/]
- 110. Op-ed: The dangers of cloning [https://funginstitute.berkeley.edu/news/op-ed-the-dangers-of-cloning/]
- 111. CRISPR Delivers RNA to Repair Neurons Right Where It's ... [https://neurosciencenews.com/crispr-rna-neural-repair-29907/]
- 112. Palmitoylation: an emerging therapeutic target bridging ... [https://cmbl.biomedcentral.com/articles/10.1186/s11658-025-00776-w]
- 113. Molecular Cloning Technology - Past, Present and Future [https://www.neb.com/en-us/tools-and-resources/feature-articles/foundations-of-molecular-cloning-past-present-and-future?srsltid=AfmBOop4iKF8rZSyLmjl7ktAJnOqd1oWSzIdcBU76G4YrCfBUwZcWJrT]
- 114. Temporal and protein-specific S-palmitoylation supports ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12515174/]
- 115. Advances in large-scale electrophysiology with high-density ... [https://pubs.rsc.org/en/content/articlehtml/2025/lc/d5lc00058k]
- 116. Nonclinical Human Neural New Approach Methodologies ... [https://www.sciencedirect.com/science/article/pii/S3050620425000594]
- 117. Top 6 Visualizations for Quantitative Data Analysis Methods [https://chartexpo.com/blog/quantitative-data-analysis-methods]
- 118. 14 Comparing quantitative data between individuals [https://bookdown.org/pkaldunn/SRM-Textbook/BetweenQuantData.html]
- 119. In Vitro Validation of the Therapeutic Potential ... [https://pubmed.ncbi.nlm.nih.gov/35628503/]
- 120. Guide to Research Techniques in Neuroscience [https://www.sciencedirect.com/book/monograph/9780128005118/guide-to-research-techniques-in-neuroscience]
- 121. Human-based complex in vitro models: their promise and ... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2025.1526306/full]