Developing Robust Semi-High-Throughput Helicase Activity Assays: A Guide for Screening and Drug Discovery
This article provides a comprehensive guide for researchers and drug development professionals on establishing semi-high-throughput screening (HTS) assays for helicase activity.
Developing Robust Semi-High-Throughput Helicase Activity Assays: A Guide for Screening and Drug Discovery
Abstract
This article provides a comprehensive guide for researchers and drug development professionals on establishing semi-high-throughput screening (HTS) assays for helicase activity. It covers the foundational principles of helicase biology and their therapeutic relevance in oncology and antiviral research. The content explores established methodological approaches, including homogeneous fluorescence-based displacement and ADP detection assays, highlighting their application in targeted campaigns against targets like WRN, SARS-CoV-2 nsp13, and DDX41. The guide also details critical troubleshooting and optimization strategies to achieve robust performance (Z' ≥ 0.7) and outlines rigorous validation workflows incorporating orthogonal and counter-screening assays to confirm mechanistic inhibition and minimize false positives.
Helicase Biology and Therapeutic Relevance: Establishing the 'Why' Behind Screening
Helicases are a class of motor enzymes that are vital to all organisms, playing indispensable roles in nearly every aspect of nucleic acid metabolism. These enzymes function as molecular motors that utilize the energy gained from ATP hydrolysis to directionally move along nucleic acid duplexes and separate hybridized strands, a process essential for accessing genetic information [1]. Approximately 1% of eukaryotic genes code for helicases, highlighting their fundamental importance to cellular function [1]. The human genome alone encodes 95 non-redundant helicases, comprising 64 RNA helicases and 31 DNA helicases, each specialized for particular pathways and functions [1].
These enzymes catalyze the unwinding of double-stranded DNA and RNA through a process characterized by the breaking of hydrogen bonds between annealed nucleotide bases [1]. Beyond their canonical unwinding activity, helicases also function to remove nucleic acid-associated proteins and catalyze homologous DNA recombination [1]. Their activities are critical for fundamental processes including DNA replication, transcription, translation, recombination, DNA repair, ribosome biogenesis, and RNA splicing, transport, editing, and degradation [1]. Some specialized helicases have also evolved to sense viral nucleic acids during infection, thereby fulfilling important immunological functions [1].
Core Biological Functions of Helicases
DNA Replication
In DNA replication, helicases serve as the fundamental unwinding engines that initiate and sustain the duplication of genetic material. These enzymes function at the replication fork, where they separate the parental double-stranded DNA into single strands, providing the templates necessary for DNA polymerase to synthesize new complementary strands [2]. This unwinding activity generates replication bubbles that enable bidirectional replication throughout the genome [2].
The replicative helicases differ between prokaryotes and eukaryotes. In prokaryotic systems, the DnaB helicase is essential for bacterial DNA replication, loaded onto DNA by DnaC initiator proteins [2]. In eukaryotic cells, the Mini-Chromosome Maintenance (MCM) complex serves as the replicative helicase, requiring additional factors such as ORC, Cdc6, and Cdt1 for proper activation and function [2]. These helicases work in coordination with single-strand DNA-binding proteins (SSBs) and replication factors to prevent reannealing and ensure efficient replication progression [2].
A critical aspect of helicase function in replication is their coupling with DNA polymerases. While isolated replicative helicases are often poor at unwinding duplex DNA on their own, and DNA polymerases cannot catalyze processive strand displacement synthesis independently, their functional and physical coupling creates a highly efficient replication machinery [3]. This partnership increases replication rate and processivity, eliminates nucleotide sequence dependency, and prevents rezipping of the unwound DNA [3]. In bacteriophage T7, for instance, this coupling is achieved through physical interactions between the C-terminal domain of the helicase and the leading strand DNA polymerase, providing stable positioning at the fork junction that is critical for efficient strand displacement synthesis [3].
DNA Repair and Recombination
Helicases play multifaceted roles in DNA repair pathways, where they help maintain genomic integrity by facilitating access to damaged DNA sites and participating in the repair mechanisms themselves. Different helicase families specialize in distinct repair pathways, forming a comprehensive network of genomic maintenance systems.
The RecQ family helicases, including WRN (Werner syndrome ATP-dependent helicase) and BLM (Bloom syndrome helicase), are particularly important in genome stability maintenance [2]. WRN is a multifunctional enzyme that possesses both magnesium and ATP-dependent DNA-helicase activity and 3'→5' exonuclease activity towards double-stranded DNA with a 5'-overhang [4]. Mutations in the WRN gene cause Werner syndrome, a disorder characterized by premature aging and increased cancer susceptibility [2]. Similarly, BLM helicase mutations result in Bloom syndrome, which predisposes individuals to various cancers due to high genomic instability [2].
Other repair helicases include UvrD helicase, which functions in nucleotide excision repair in prokaryotes, and XPD/XPB helicases, which are involved in both nucleotide excision repair (NER) and transcription [2]. In nucleotide excision repair, these helicases help unwind DNA around damaged sites to allow excision and replacement of faulty sequences. Defects in these repair helicases can have severe consequences; XPB/XPD mutations cause Xeroderma Pigmentosum, characterized by extreme UV sensitivity and high skin cancer risk, while FANCJ helicase deficiency leads to Fanconi Anemia, resulting in bone marrow failure and increased cancer risk [2].
RNA Metabolism
RNA helicases, particularly those belonging to the DEAD-box family, facilitate various aspects of RNA metabolism through their strand separation and remodeling activities. These enzymes are involved in pre-mRNA splicing, mRNA stability and translation, ribosome biogenesis, RNA transport, and RNA degradation [1] [5]. Unlike DNA helicases that catalyze processive translocation, many DEAD-box RNA helicases perform local strand separation induced upon NTP-dependent binding to RNA with limited NTP hydrolysis required for enzyme recycling [6].
The DDX3 subfamily of DEAD-box RNA helicases exemplifies the diverse roles of RNA helicases in cellular function. Humans express two DDX3 homologs: DDX3X, encoded on the X-chromosome and ubiquitously expressed in all tissues, and DDX3Y, encoded on the Y-chromosome with protein expression restricted to the testis where it plays an essential role in spermatogenesis and male fertility [5]. These helicases share approximately 92% amino acid sequence similarity and play indispensable, often compensatory roles in various cellular processes [5]. They regulate a wide range of biological functions, including cell adhesion, cell cycle progression, and cellular stress responses [5].
Pathogenic mutations in RNA helicase genes are associated with multiple human diseases. Germline inheritance of pathogenic DDX3X mutations hinders neurodevelopment, accounting for approximately 1-3% of cases with intellectual disability [5]. In cancer, DDX3X can function as either a tumor suppressor or an oncogene depending on tumor type, with overexpression observed in breast, colorectal, lung, medulloblastoma, and prostate cancers, and somatic mutations found in medulloblastoma, melanoma, and non-Hodgkin lymphoma subtypes [5].
Immune Signaling
Specialized helicases participate in immune signaling pathways by sensing viral nucleic acids during infection and initiating appropriate immune responses. These helicases function as pattern recognition receptors that detect foreign nucleic acid patterns and trigger signaling cascades leading to interferon production and antiviral defense mechanisms.
While the search results provide limited specific details about individual immune helicases, they confirm that some helicases fulfill immunological functions by sensing viral nucleic acids during infection [1]. These helicases likely contribute to the distinction between self and non-self nucleic acids, a crucial aspect of antiviral immunity, and may be involved in pathways that detect viral replication intermediates or unusual nucleic acid structures associated with infection.
Table 1: Key Helicase Functions in Cellular Pathways
| Cellular Pathway | Representative Helicases | Primary Functions | Associated Diseases |
|---|---|---|---|
| DNA Replication | DnaB (prokaryotic), MCM complex (eukaryotic) | Unwinds DNA at replication fork, coordinates with DNA polymerase | Cancer, developmental disorders |
| DNA Repair | WRN, BLM, XPD, XPB, UvrD, FANCJ | Nucleotide excision repair, homologous recombination, mismatch repair | Werner syndrome, Bloom syndrome, Xeroderma pigmentosum, Fanconi anemia |
| RNA Metabolism | DDX3X, DDX3Y, other DEAD-box proteins | pre-mRNA splicing, translation initiation, ribosome biogenesis, RNA transport | Intellectual disability, cancer, male infertility |
| Immune Signaling | Specialized sensor helicases | Viral nucleic acid detection, interferon pathway activation | Immunodeficiency, autoimmune disorders |
Quantitative Analysis of Helicase Activities
Understanding the kinetic parameters and functional characteristics of helicases is essential for both basic research and drug discovery efforts. The quantitative profiling of helicase activities provides insights into their mechanisms and facilitates the development of targeted therapeutics.
Table 2: Kinetic Parameters of Selected SF1 Helicases
| Helicase | Organism | Directionality | Unwinding Rate (bp/s) | Processivity (bp) | Cellular Functions |
|---|---|---|---|---|---|
| UvrD | Escherichia coli | 3'→5' | 250 | 240 | DNA repair |
| Rep | Escherichia coli | 3'→5' | 45 | 30 | DNA replication |
| PcrA | Bacillus stearothermophilus | 3'→5' | 31 | 5.5 | DNA repair, plasmid replication |
| TraI | Escherichia coli | 5'→3' | 1120 | >850 | DNA transfer during conjugation |
| Pif1 | Saccharomyces cerevisiae | 5'→3' | 75 | 10 | Mitochondrial DNA maintenance |
| Dda | phage T4 | 5'→3' | 262 | 64 | DNA replication initiation, recombination |
| Upf1 | Homo sapiens | 5'→3' | 0.16-0.32 (RNA) | >10^4 | Telomere maintenance, mRNA decay |
The quantitative analysis of helicase activities reveals several important patterns. Helicases exhibit significant variation in their unwinding rates, spanning from less than 1 bp/s to over 1000 bp/s, reflecting their adaptation to specific cellular roles [7]. Similarly, processivity values range from just a few base pairs to thousands, with some RNA helicases like Upf1 demonstrating particularly high processivity [7]. The directionality of unwinding is also a key functional parameter, with SF1 helicases divided into SF1A (3'→5') and SF1B (5'→3') categories based on their translocation polarity [7].
The energy requirements for helicase function are substantial. Under physiological conditions, the energy of NTP hydrolysis is approximately 12.1 kcal/mole or ~20 kBT, which translates to 86 pN-nm at 310K [6]. For a maximally efficient helicase, unwinding 3-10 bp (moving ~1-3.4 nm) translates into enzyme force generation of 25-86 pN [6]. These estimates align with experimental measurements of 10-20 pN force required to unzip nucleic acids in the absence of enzymes, providing a theoretical framework for the physical parameters of helicase operation [6].
Experimental Protocols for Helicase Activity Analysis
Molecular Beacon-Based Helicase Assay
The molecular beacon-based helicase assay provides a continuous, fluorescence-based method for monitoring helicase activity in real time without requiring separation of reaction products [8]. This assay utilizes a single-stranded DNA oligonucleotide molecular beacon featuring a fluorescent moiety attached to one end and a quencher attached to the other, annealed to a longer DNA or RNA oligonucleotide [8].
Protocol Steps:
- Substrate Preparation: Combine single strands at a 1:1 molar ratio to a final concentration of 20 μM in 10 mM Tris HCl pH 8.5. Heat to 95°C in a water bath, then allow to cool slowly to room temperature for approximately 1 hour to facilitate proper annealing [8].
Reaction Setup: Prepare reaction mixtures containing:
- 25 mM MOPS pH 6.5
- 2 mM MgCl₂
- 25 nM helicase enzyme
- 5 nM molecular beacon substrate
- Reactions are typically carried out in 100 μL volumes in white "half-volume" 96-well polystyrene plates [8].
Reaction Initiation and Monitoring: Initiate the reaction by adding ATP to a final concentration of 0.5 mM. Monitor fluorescence continuously every 40 seconds using a fluorescence spectrophotometer equipped with a microplate reader [8]. For Cy3-labeled substrates, measure excitation/emission at 552/570 nm; for Cy5-labeled substrates, use 643/667 nm [8].
Data Analysis: Analyze data using appropriate software, applying a first-order exponential decay model to determine the pseudo-first order rate constant (kobs) [8].
Advantages and Considerations: The molecular beacon assay offers several advantages over traditional methods. It is continuous and irreversible due to intramolecular hairpin formation that prevents strand reannealing, eliminating the need for single-stranded DNA trap molecules [8]. The design minimizes potential impact on observed reaction rates by contacting primarily one strand of the duplex, and the assay is amenable to high-throughput screening applications [8]. This method has been successfully validated using HCV NS3 helicase as a model system [8].
ADP-Glo Max Assay for ATPase Activity
The ADP-Glo Max Assay provides a luminescence-based method for quantifying ATPase activity, which directly correlates with helicase function since helicases couple ATP hydrolysis to mechanical work on nucleic acids [9]. This assay is particularly suitable for high-throughput screening of helicase inhibitors or activators.
Protocol Steps:
- Enzyme Reaction Setup:
ADP Detection:
- Add an equal volume of ADP-Glo Reagent to terminate the reaction and deplete remaining ATP.
- Incubate for 40-60 minutes at room temperature.
- Add Kinase Detection Reagent to convert ADP to ATP while simultaneously generating light from the newly synthesized ATP.
- Incubate for 30-60 minutes at room temperature [9].
Signal Measurement:
- Measure luminescence using a compatible plate reader.
- The luminescent signal is directly proportional to the amount of ADP produced, which correlates with helicase ATPase activity [9].
Applications and Validation: This assay has been extensively validated for high-throughput screening and inhibitor dose response measurements [4]. It has been successfully applied to characterize WRN helicase inhibitors, including the identification of benzimidazole analogs that reduced the IC₅₀ for WRN ATPase inhibition from 88 nM to 5 nM [9]. The assay has also been used to study Pol θ ATPase inhibitors like novobiocin, confirming non-competitive inhibition through 14-point ADP-Glo assays [9].
In-Cell DDX3 Helicase (ICD-Helicase) Reporter Assay
The ICD-helicase reporter system represents an innovative cell-based approach for evaluating helicase activities in a biologically relevant context that accounts for cellular complexity and signaling pathways [5]. This system addresses limitations of conventional cell-free assays by maintaining physiological conditions.
Protocol Steps:
- Reporter Cell Line Generation:
- Create DDX3X knockout 293T cells using CRISPR/Cas9 technology with sgRNAs targeting DDX3X sequences (e.g., CGTGGACGGAGTGATTACGA) [5].
- Co-transfect with plasmids expressing sgRNA against DDX3X and the heparin-binding EGF-like growth factor (HBEGF) for selection.
- Treat with 20 ng/mL diphtheria toxin to enrich for knockout cells, then select single cell clones to establish stable DDX3X KO cells [5].
Reporter Assay Execution:
- Transfect DDX3X KO cells with firefly luciferase plasmids that provide bioluminescence signals dependent on helicase activities of exogenously expressed wild-type or mutant DDX3X or DDX3Y.
- Include Aequorea coerulescens Green Fluorescent Protein (AcGFP) as an internal control separated by an internal ribosome entry site (IRES) [5].
- Culture transfected cells for 24-48 hours to allow protein expression.
Activity Measurement:
- Lyse cells in luciferase cell lysis buffer.
- Measure bioluminescence in white bottom 96-well plates using a luciferase assay system according to manufacturer instructions.
- Analyze the other half of cells by flow cytometry to determine the percentage of AcGFP+ cells for normalization against transfection efficiency [5].
Applications and Advantages: The ICD-helicase reporter system enables functional interrogation of DDX3X and DDX3Y helicase activities and their mutational variants in living cells [5]. This system can be applied to screen compound libraries targeting DDX3X or DDX3Y, which are implicated in cancer and several other diseases, and to study their functional roles in health and disease [5]. The cellular context preserves native interactions, post-translational modifications, and compartmentalization that may influence helicase function.
Research Reagent Solutions
Table 3: Essential Research Reagents for Helicase Studies
| Reagent Category | Specific Examples | Function and Applications |
|---|---|---|
| Helicase Enzymes | Purified human WRN helicase (amino acids 500-946, N-terminal 6xHis) [4] | Biochemical assays, inhibitor screening, kinetic studies |
| DNA Substrates | 37-bp annealed 3'-Flap duplex DNA oligomer [4], Molecular beacon substrates [8] | Unwinding assays, mechanism studies, high-throughput screening |
| Assay Buffer Systems | Enzyme Assay Buffer A (500 mM TRIS pH 7.5, 10 mM MgCl₂, 0.1% Triton) [4] | Optimized reaction conditions for helicase activity |
| Detection Reagents | Transcreener ADP2 Assay Kits (FP, FI, TR-FRET formats) [4], ADP-Glo Max Assay [9] | ATPase activity measurement, compound screening |
| Cell-Based Reporter Systems | ICD-helicase reporter constructs (5'UTR-luciferase pIRES2-AcGFP1) [5] | In-cell helicase activity assessment, pathway analysis |
| Inhibitors and Modulators | RK-33 (DDX3 inhibitor) [5], Novobiocin (Pol θ inhibitor) [9], HRO761 (WRN inhibitor) [9] | Mechanistic studies, target validation, therapeutic development |
Experimental Workflows and Signaling Pathways
Molecular Beacon Helicase Assay Workflow
ATPase-Coupled Helicase Function Diagram
DNA Replication Fork with Helicase-Polymerase Coupling
Helicases as High-Value Drug Targets in Cancer and Antiviral Therapy
Helicases are ubiquitous molecular motor proteins that utilize the energy from adenosine triphosphate (ATP) hydrolysis to unwind double-stranded DNA and RNA, separate nucleic acid secondary structures, and remodel nucleoprotein complexes. These functions are essential for virtually all aspects of nucleic acid metabolism, including DNA replication, repair, recombination, transcription, RNA processing, and translation [10] [11]. The critical roles of helicases in maintaining genomic integrity and facilitating gene expression make them attractive therapeutic targets for cancer and viral infections. In cancer cells, dysregulation of DNA repair helicases can create unique vulnerabilities, while viral replication often depends on specific virally-encoded helicases not found in host cells [12] [10] [13].
Germline mutations in several DNA repair helicases are implicated in human genetic disorders characterized by genomic instability, cancer predisposition, and premature aging. These include Werner syndrome (WRN), Bloom syndrome (BLM), Rothmund–Thomson syndrome (RECQL4), and Xeroderma pigmentosum (XPB/XPD) [12]. From a therapeutic perspective, this dependency creates opportunities for synthetic lethality approaches, where inhibition of a backup DNA repair pathway selectively kills cancer cells already deficient in a specific helicase function. In antiviral therapy, targeting essential viral helicases or host helicases co-opted by viruses offers strategies to suppress viral replication across diverse pathogens, including coronaviruses, hepatitis C virus (HCV), and herpes simplex virus (HSV) [14] [10] [13].
Therapeutic Significance of Key Helicase Targets
DNA Repair Helicases in Oncology
Table 1: Key DNA Repair Helicases as Anticancer Targets
| Helicase | Classification | Primary Functions | Associated Diseases | Therapeutic Rationale | Reported Inhibitors |
|---|---|---|---|---|---|
| WRN | SF2 (RecQ family) | DNA repair, telomere maintenance, replication fork restart | Werner syndrome, aging, cancer | Synthetic lethality with microsatellite instability (MSI) | Under investigation [4] |
| BLM | SF2 (RecQ family) | DNA unwinding, genome stability, replication fork restart | Bloom syndrome, cancer predisposition | Synthetic lethality in homologous repair-deficient cancers | Under investigation [15] |
| PIF1 | SF1B | G-quadruplex resolution, telomere maintenance, mitochondrial DNA repair | Cancer (upregulated in tumors) | Oncogene-driven replication stress creates dependency | 4-phenylthiazol-2-amine derivatives [16] |
| XPB | SF2 | Component of TFIIH, DNA opening in NER | Xeroderma Pigmentosum (XP), Trichothiodystrophy (TTD) | Inhibition sensitizes cancer cells to cisplatin and other DNA-damaging agents | Triptolide, Minnelide, Spironolactone [12] |
Dysfunctional helicases are directly implicated in oncogenesis and cancer cell survival. For example, the RecQ family helicases WRN and BLM act as tumor suppressors; their loss causes Werner and Bloom syndromes, respectively, which are characterized by cancer predisposition and genomic instability [12]. Therapeutically, WRN has emerged as a promising synthetic lethal target in cancers with microsatellite instability (MSI). Similarly, BLM inhibition is being explored for selective targeting of homologous recombination-deficient cancers [15]. Beyond the RecQ family, helicases like PIF1 are upregulated in certain tumors, where they help resolve oncogene-induced replication stress, such as stabilizing G-quadruplex structures. Recent research has identified the first inhibitors targeting the helicase activity of human PIF1, providing a foundation for a novel class of anticancer therapeutics [16].
Nucleotide Excision Repair (NER) helicases also present attractive targets for chemo-sensitization. XPB, a subunit of the transcription factor IIH (TFIIH) complex, is essential for the DNA unwinding step in NER. Inhibiting XPB can disrupt the repair of DNA crosslinks caused by chemotherapeutic agents like cisplatin, thereby sensitizing cancer cells to these treatments. The natural compound triptolide and its water-soluble derivative minnelide (which has advanced to clinical trials) covalently bind and inhibit XPB's ATPase activity. The diuretic spironolactone has also been repurposed to degrade XPB and enhance the efficacy of platinum-based chemotherapies in preclinical models of bladder cancer and glioblastoma [12].
Viral and Host Helicases in Antiviral Therapy
Table 2: Key Viral and Host Helicases as Antiviral Targets
| Helicase | Origin | Virus/Function | Therapeutic Rationale | Inhibitor Status |
|---|---|---|---|---|
| nsP13 | Viral | SARS-CoV-2 RNA replication | Essential for viral replication; direct antiviral target | HTS identified 674 compounds with IC50 <10 μM [14] [17] |
| NS3 | Viral | Hepatitis C Virus (HCV) RNA replication | Essential for viral replication; direct antiviral target | Multiple chemotypes identified [10] [13] |
| DDX3 | Host (Human) | Cofactor for HIV-1 and other viruses | Broad-spectrum potential; higher barrier to viral resistance | Under investigation [13] [15] |
| RIG-I (DDX58) | Host (Human) | Cytosolic RNA sensor activating interferon | Immunotherapy target for antiviral and oncology | Assays available for inhibitor/modulator screening [15] |
Targeting helicases has emerged as a viable antiviral strategy with two primary approaches: inhibiting virally-encoded helicases or targeting host helicases that viruses hijack for their replication. SARS-CoV-2 non-structural protein 13 (nsP13) is an RNA helicase essential for viral replication. Recent high-throughput screening (HTS) of approximately 650,000 compounds identified 674 hits with inhibitory activity (IC50 <10 μM) against nsP13, demonstrating the druggability of this target and providing a pipeline for future antiviral development [14] [17]. Similarly, the HCV NS3 helicase has been extensively studied, and while discovering specific inhibitors has been challenging, it remains a validated target for anti-HCV therapy [10] [13].
Targeting host helicases required for viral replication offers a complementary strategy with a potentially higher genetic barrier to resistance. For instance, the human DEAD-box RNA helicase DDX3 is exploited by multiple viruses, including HIV-1, HCV, and dengue virus, for various steps in their life cycles. Inhibiting DDX3 could, therefore, yield broad-spectrum antivirals. However, this approach requires exquisite selectivity to avoid toxicity from disrupting the essential cellular functions of these helicases [13] [15]. Other host helicases like RIG-I (DDX58) and MDA5 are cytosolic sensors that activate interferon responses upon viral RNA detection; modulating their activity represents an immunotherapeutic approach to antiviral treatment [15].
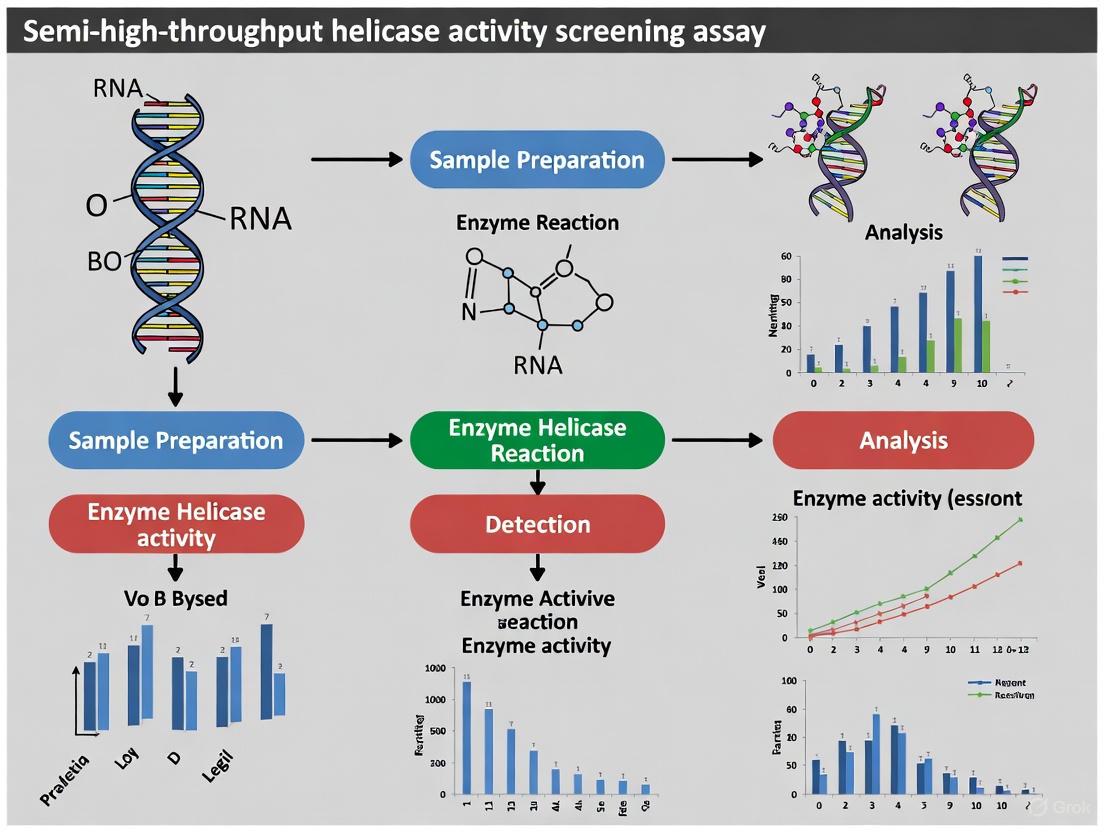
Experimental Approaches for Helicase Activity Screening
Selecting an appropriate assay is critical for successful helicase inhibitor screening. The ideal assay combines high sensitivity, robustness, throughput, and biological relevance. The table below compares the major biochemical formats used to detect helicase activity, highlighting their principles and applications [15].
Table 3: Major Biochemical Assay Formats for Helicase Activity Screening
| Assay Format | Readout Principle | Advantages | Limitations | Best Use Cases |
|---|---|---|---|---|
| Gel-based Unwinding | Separation of labeled duplex/unwound strands via electrophoresis. | Direct visualization; considered a gold standard for validation. | Low throughput; labor-intensive and time-consuming. | Mechanistic validation and follow-up studies. |
| Fluorescent Dye Displacement | Fluorescence decrease as intercalating dye is released during unwinding. | Continuous monitoring; simple setup. | Dye may perturb duplex; potential compound interference. | General kinetic studies. |
| Heliscreener-type Unwinding | Fluorescence increase as quencher and fluorophore are separated. | Real-time, high-throughput, highly sensitive. | Requires carefully optimized substrate design. | Primary HTS, kinetics, inhibitor profiling. |
| ADP Detection (e.g., Transcreener ADP²) | Detects ADP produced from ATP hydrolysis. | Universal, homogeneous, HTS-ready, robust. | Indirect (measures ATPase activity only). | Primary HTS, broad applicability. |
| 2-Aminopurine Incorporation | Fluorescence increase as the 2-AP base is unpaired. | Minimal substrate labeling. | Low signal change (~2-fold). | Mechanistic follow-up. |
For high-throughput screening (HTS) of compound libraries, homogeneous "mix-and-read" assays are preferred. Two of the most robust formats are the Heliscreener-type unwinding assays, which directly measure strand separation, and ADP detection assays, which indirectly monitor helicase activity via ATP hydrolysis [15]. The Transcreener ADP² assay, for instance, is a far-red, competitive fluorescence immunoassay that enables single-addition, mix-and-read detection in continuous or endpoint formats. It has been extensively validated for HTS and inhibitor dose-response measurements, typically yielding robust Z′ factors ≥ 0.7 [4] [15]. A recommended workflow involves primary screening using a universal ADP detection assay for cost-efficiency, followed by orthogonal confirmation of hits using a direct unwinding assay to rule out artifacts and confirm mechanistic activity [15].
Detailed Protocol: WRN Helicase ATPase Assay System
The following protocol utilizes the Enzolution WRN Helicase ATPase Assay System, intended for use with Transcreener ADP2 Assay Kits to measure the enzymatic activity of WRN helicase, a target in cancer therapy [4].
Principle
WRN is a multifunctional enzyme with ATP-dependent helicase activity. This assay quantifies ADP formation resulting from WRN's ATPase activity, which is coupled to its helicase function. The Transcreener ADP2 assay detects ADP using a competitive fluorescence polarization (FP), fluorescence intensity (FI), or time-resolved FRET (TR-FRET) format between an ADP-specific antibody and a fluorescent ADP tracer [4].
Materials Provided
- WRN Helicase Enzyme: Purified human WRN (amino acids 500-946, N-terminal 6xHis), 0.1 mg/mL (≈1.93 µM) in storage buffer.
- WRN-H DNA Substrate: 40 µM annealed 37-bp 3’-Flap duplex DNA oligomer in H₂O.
- Enzyme Assay Buffer A, 10X: 500 mM Tris (pH 7.5), 10 mM MgCl₂, 0.1% Triton.
- 384-Well Low Volume Assay Plates (e.g., Corning #4514 for FP/FI).
Materials required but not provided: Transcreener ADP2 Assay Kit (antibody, tracer, Stop & Detect Buffer), ultrapure nuclease-free water, a compatible multimode plate reader, and liquid handling devices [4].
Procedure
Reaction Setup:
- Dilute the 10X Enzyme Assay Buffer A to 1X in nuclease-free water.
- Prepare a master mix containing 1X Assay Buffer, WRN Helicase (final concentration ~50-100 nM), and DNA substrate (final concentration ~200-500 nM).
- Dispense the enzyme/master mix into the 384-well assay plate. Pre-incubate with inhibitors for 15-30 minutes.
Initiate Enzyme Reaction:
- Start the reaction by adding ATP (from the Transcreener kit) to a final concentration of 50-100 µM. The final reaction volume is 10 µL.
- Incubate the plate at 30°C for 30-60 minutes to allow ADP formation.
Stop Reaction and Detect ADP:
- Stop the reaction by adding 10 µL of Stop & Detect Buffer (containing EDTA to chelate Mg²⁺ and the fluorescent tracer).
- Add the ADP-specific antibody (volume per Transcreener kit instructions).
- Incubate the plate at room temperature for 30-60 minutes.
Plate Reading and Data Analysis:
- Read the plate using the appropriate mode on the plate reader (FP, FI, or TR-FRET).
- Calculate the percentage of inhibition for test compounds using positive (no inhibitor) and negative (no enzyme) controls.
- Generate dose-response curves and calculate IC₅₀ values for confirmed hits [4].
The Scientist's Toolkit: Key Research Reagent Solutions
Table 4: Essential Research Tools for Helicase Inhibitor Discovery
| Tool / Resource Name | Type | Function / Application | Example / Source |
|---|---|---|---|
| Transcreener ADP2 Assay | Biochemical Kit | Universal, HTS-ready detection of ADP formation from ATP hydrolysis by helicases. | BellBrook Labs [4] [15] |
| Heliscreener Platform | Biochemical Assay | Direct, real-time measurement of nucleic acid strand displacement/unwinding. | BellBrook Labs [15] |
| Enzolution WRN System | Enzyme/Substrate System | Provides purified WRN helicase and optimized DNA substrate for targeted screening. | BellBrook Labs [4] |
| Heli-SMACC Database | Bioinformatics Database | Curated collection of >13,500 molecules tested against 29 helicases; aids in hit identification. | https://smacc.mml.unc.edu [18] |
| Purified Helicases (BLM, POLQ, DDX3, etc.) | Recombinant Proteins | Essential for biochemical assays and profiling inhibitor selectivity. | Commercial vendors (e.g., BellBrook Labs [15]) |
Visualizing Pathways and Workflows
Therapeutic Targeting Pathways
The following diagram illustrates the key roles of helicases in cancer and viral infection, highlighting potential intervention points for therapeutic inhibitors.
High-Throughput Screening Workflow
This diagram outlines a generalized semi-high-throughput workflow for the discovery and validation of helicase inhibitors.
Helicases are essential enzymes that unwind nucleic acid duplexes, playing critical roles in DNA replication, repair, and RNA metabolism. Their fundamental functions make them attractive targets for therapeutic intervention in diseases ranging from cancer to viral infections. This application note focuses on two prominent helicase targets: human Werner syndrome helicase (WRN) for microsatellite instable (MSI) cancers and viral SARS-CoV-2 nonstructural protein 13 (nsp13) for antiviral development.
The discovery of synthetic lethality between WRN and MSI cancers has established WRN as a promising target in oncology [19] [20]. Simultaneously, the essential role of nsp13 in SARS-CoV-2 replication and its high conservation among coronaviruses position it as a valuable target for broad-spectrum antiviral development [14] [21]. This note provides detailed methodologies for screening and characterizing inhibitors of these therapeutically relevant helicases, supporting drug discovery efforts in both fields.
Therapeutic Target Profiles and Biological Significance
Table 1: Key Characteristics of WRN and SARS-CoV-2 nsp13 Helicases
| Feature | WRN Helicase | SARS-CoV-2 nsp13 |
|---|---|---|
| Primary Therapeutic Area | Oncology (MSI Cancers) | Antiviral (COVID-19/Treatment) |
| Biological Role | Genome integrity, DNA repair, replication | Viral RNA replication & transcription |
| Significance | Synthetic lethal target in MSI-H/dMMR cancers | Essential for viral replication complex |
| Dependency | MSI tumor cells are dependent on WRN for survival | Coronaviruses are dependent on nsp13 for replication |
| Key Structural Features | RecQ helicase family; unique exonuclease domain | SF1B helicase; Zinc-binding domain (ZBD) |
| Catalytic Activities | 3'→5' DNA helicase, 3'→5' exonuclease | 5'→3' RNA/DNA helicase, NTP hydrolysis |
| Conservation | Human RecQ family (5 members) | Highly conserved across coronaviruses |
WRN in Microsatellite Instable Cancers
Microsatellite instability (MSI) occurs in cancers with deficient DNA mismatch repair (dMMR) and is present in subsets of colorectal, gastric, endometrial, and other cancers [19]. In 2019, multiple independent genetic screens identified WRN as a synthetic lethal target in MSI cancer models [20]. MSI cancer cells accumulate numerous insertion/deletion mutations in repetitive DNA sequences, leading to DNA secondary structures that require WRN's helicase activity for resolution during replication [19] [22]. When WRN is inhibited, these structures persist, causing DNA double-strand breaks, cell cycle arrest, and apoptosis specifically in MSI cells, while microsatellite stable (MSS) cells remain unaffected [19] [22] [20]. This selective dependency makes WRN an attractive target for precision oncology approaches.
Clinical-stage WRN inhibitors like HRO761 (Novartis) bind allosterically at the D1-D2 helicase domain interface, locking WRN in an inactive conformation and recapitulating the synthetic lethal effect observed with genetic suppression [22]. In preclinical models, HRO761 treatment resulted in dose-dependent DNA damage induction and tumor growth inhibition in MSI cell line-derived and patient-derived xenografts, providing pharmacological validation of WRN targeting [22].
SARS-CoV-2 nsp13 Helicase in Antiviral Therapy
SARS-CoV-2 nsp13 is a superfamily 1B (SF1B) helicase that is part of the viral replication-transcription complex (RTC) and is essential for viral replication [21]. The enzyme possesses 5' to 3' unwinding activity on double-stranded RNA and DNA, along with RNA 5' triphosphatase activity believed to be involved in viral mRNA capping [21]. Its high conservation (99.8% sequence identity with SARS-CoV-1) and essential role make it an attractive target for developing broad-spectrum coronavirus inhibitors [23] [21].
Structural studies have revealed that nsp13 consists of five domains: a zinc-binding domain (ZBD), a stalk domain, a 1B domain, and two RecA-like domains (1A and 2A) that form the helicase core [21]. The presence of multiple druggable pockets, including the nucleotide-binding site and RNA-binding channel, provides opportunities for therapeutic intervention [23] [21]. While no FDA-approved nsp13 inhibitors currently exist, several candidate molecules have been identified through screening campaigns, including IOWH-032, which inhibits both ATPase and helicase activities at low micromolar concentrations by interacting with the RNA-binding interface [23].
Quantitative Profiling of Helicase Targets and Inhibitors
Table 2: Quantitative Profile of Helicase Inhibitors in Development
| Inhibitor / Molecule | Target | Biochemical IC₅₀ | Cellular IC₅₀ / GI₅₀ | Mechanism of Action | Development Status |
|---|---|---|---|---|---|
| HRO761 (Novartis) | WRN | ~100 nM (ATPase) | 40 - 1,000 nM (MSI cells) | Allosteric inhibitor; induces inactive conformation & degradation | Phase I Clinical Trial (NCT05838768) [22] |
| H3B-219 | WRN | Nanomolar range | Not Specified | Covalent inhibitor targeting C727 [24] | Preclinical [24] |
| VVD-133214 | WRN | Not Specified | Not Specified | Covalent inhibitor targeting C727 [24] | Phase I Clinical Trial (NCT06004245) [24] |
| IOWH-032 | nsp13 | 28.3 μM (ATPase) | Low micromolar (viral load reduction) | Binds RNA interface, displaces nucleic acid substrate [23] | Repurposed candidate; Preclinical for SARS-CoV-2 [23] |
| SSYA10-001 | nsp13 | Low micromolar | Antiviral activity in cells | Inhibits helicase activity [23] | Preclinical for SARS-CoV-1/2 [23] |
Experimental Protocols for Helicase Activity and Inhibition Screening
Protocol: High-Throughput Screening for WRN Helicase Inhibitors
Principle: This fluorometric assay detects WRN helicase activity through the unwinding of a forked duplex DNA substrate with fluorescence resonance energy transfer (FRET) pair. When the strands are annealed, the fluorophore (TAMRA) is quenched by BHQ2. Unwinding separates the strands, increasing fluorescence [25].
Reagents:
- Recombinant WRN helicase fragment (GST-WRN500-946) or full-length WRN
- Forked DNA substrate: OLIGOA-BHQ2 and OLIGOB-TAMRA
- Assay Buffer: 50 mM Tris-HCl (pH 8.0), 5 mM MgCl₂, 100 mM NaCl, 0.01% Triton X-100, 1 mM DTT
- ATP solution (100 μM final concentration)
- Test compounds in DMSO (final DMSO concentration ≤1%)
Procedure:
- Substrate Preparation: Anneal equal amounts of OLIGOA-BHQ2 and OLIGOB-TAMRA in annealing buffer (50 mM NaCl) by heating to 95°C for 5 minutes and slowly cooling to room temperature.
- Reaction Setup: In 384-well plates, add:
- 20 nM FORKF DNA substrate
- 10 nM WRN enzyme
- Test compounds (various concentrations)
- ATP (100 μM final concentration) in assay buffer
- Control Wells Include:
- No enzyme control (background signal)
- No ATP control (background signal)
- DMSO vehicle control (maximum activity)
- Reference inhibitor control if available
- Incubation: Incubate at 30°C for 60 minutes.
- Detection: Measure fluorescence (TAMRA excitation/emission: 555/580 nm) using a plate reader.
- Data Analysis: Calculate % inhibition = [1 - (Fsample - Fbackground)/(Fcontrol - Fbackground)] × 100
Validation: The assay demonstrated robustness with Z' factor >0.8, suitable for high-throughput screening of compound libraries [25].
Protocol: SARS-CoV-2 nsp13 Helicase Unwinding Assay
Principle: This FRET-based assay measures nsp13's 5'→3' unwinding activity using a DNA substrate with Cy3 fluorophore on one strand and BHQ2 quencher on the complementary strand. Unwinding increases fluorescence as the quencher separates from the fluorophore [23] [26].
Reagents:
- Purified SARS-CoV-2 nsp13 protein
- DNA substrate: Cy3-labeled strand with complementary BHQ2-labeled strand
- Reaction Buffer: 50 mM HEPES (pH 7.5), 5 mM MgCl₂, 1 mM DTT, 0.01% Triton X-100
- ATP solution (1 mM final concentration)
- Test compounds
Procedure:
- Substrate Preparation: Anneal Cy3- and BHQ2-labeled strands in equimolar ratios.
- Reaction Setup: In 384-well plates, add:
- 50 nM DNA substrate
- 100 nM nsp13
- Test compounds (various concentrations)
- ATP (1 mM final) in reaction buffer
- Kinetic Measurement: Monitor fluorescence (Cy3 excitation/emission: 550/570 nm) continuously for 30-60 minutes at 30°C.
- Data Analysis: Calculate initial velocities from linear phase and determine IC₅₀ values from dose-response curves.
Notes: Although nsp13's natural substrate is RNA, DNA substrates provide greater stability and generate more robust signals in vitro [23]. The ATPase activity of nsp13 can be measured in parallel using ADP-Glo or similar coupled assays for compound characterization [26].
Signaling Pathways and Experimental Workflows
Diagram 1: Mechanism of Action for WRN and nsp13 Helicase Inhibitors. WRN inhibition exploits synthetic lethality in MSI cancers, while nsp13 targeting disrupts viral replication.
Diagram 2: Integrated Workflow for Helicase Inhibitor Discovery. The process spans from initial assay development through lead optimization, utilizing multiple orthogonal methods for compound validation.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagent Solutions for Helicase Studies
| Reagent / Material | Function / Application | Example Specifications |
|---|---|---|
| Transcreener ADP² Assay Kit | HTS-compatible ADP detection for ATPase activity; used for BLM, WRN, and other helicases [27] | Homogeneous, mix-and-read format; Z' > 0.8; 96/384/1536-well compatible [27] |
| Recombinant WRN Protein | Biochemical assays and screening; catalytic domain (e.g., GST-WRN500-946) or full-length | Baculovirus or E. coli expression; helicase and exonuclease activity validation [25] |
| Recombinant nsp13 Protein | SARS-CoV-2 helicase assays and inhibitor screening | Full-length (1-601); ≥90% purity; ATPase and helicase activity confirmed [26] [21] |
| FRET DNA/RNA Substrates | Helicase unwinding activity measurement | Forked duplex DNA with fluorophore-quencher pair (e.g., Cy3/BHQ2, TAMRA/BHQ2) [23] [25] |
| Fragment Libraries | Fragment-based drug discovery against helicase targets | 500+ fragments for NMR screening; diverse chemotypes [26] |
| HTS-Compatible Assay Plates | Screening compound libraries | 384-well black plates (e.g., Corning #4514) for fluorescence-based assays [27] |
Helicases represent promising therapeutic targets with WRN inhibition offering a novel approach for MSI cancers and nsp13 targeting providing potential for broad-spectrum antivirals. The application notes and protocols detailed here provide a framework for semi-high-throughput screening of helicase inhibitors, from assay development through mechanistic characterization. As clinical validation of these targets progresses, particularly with multiple WRN inhibitors now in Phase I trials, the methodologies outlined will support continued drug discovery efforts against these biologically significant and therapeutically relevant helicase targets.
The Rationale for Semi-High-Throughput Screening in Helicase Inhibitor Discovery
Helicases are essential motor proteins that unwind nucleic acid duplexes, playing critical roles in genome replication, repair, and transcription. Their dysregulation is implicated in various diseases, including cancer, viral infections, and genetic disorders, making them attractive therapeutic targets [28] [29]. The discovery of helicase inhibitors presents unique challenges due to the complex enzyme kinetics, the necessity for multiple orthogonal assays, and the need to distinguish between specific inhibition and general nucleic acid binding [29]. Semi-high-throughput screening (semi-HTS) has emerged as a powerful strategy that bridges the gap between low-throughput mechanistic studies and ultra-HTS campaigns, offering a balanced approach for identifying and validating novel helicase inhibitors with a focus on quality and mechanistic insight. This approach is particularly valuable for targeting viral helicases such as SARS-CoV-2 nsP13, a highly conserved enzyme crucial for viral replication and a promising target for broad-spectrum antivirals [30] [26].
The Case for Semi-HTS in Helicase Drug Discovery
Addressing the Challenges of Helicase Assays
Traditional helicase inhibitor screening faces several technical hurdles. A significant problem is the prevalence of false positives, where compounds interfere with the assay readout (e.g., by quenching fluorescence) or act through non-specific mechanisms like aggregation or covalent modification of the protein [29]. Semi-HTS addresses this by incorporating multiple orthogonal assays early in the screening funnel, enabling rapid triage of false positives and confirming true mechanistic inhibitors [26]. Furthermore, the quantitative data generated in semi-HTS, such as IC50 values, allows for a more nuanced prioritization of hits compared to simple "active/inactive" classifications from primary ultra-HTS [30].
Successful Applications in Targeting Viral Helicases
The rationale for semi-HTS is strongly supported by its successful application in recent campaigns against the SARS-CoV-2 nsP13 helicase. One study implemented a robust, semi-HTS-compatible biochemical assay in a 1,536-well plate format to screen a library of approximately 650,000 compounds [30]. The primary screen was highly robust, with an average Z' factor of 0.86 ± 0.05, leading to the identification of 7,009 primary hits. Through repeated retesting and titration assays, this list was refined to 674 compounds with an IC50 of less than 10 µM, demonstrating the funnel's effectiveness [30]. In a parallel, integrated approach, a fragment-based screening campaign using NMR spectroscopy screened a 500-fragment library. This semi-HTS method identified 40 high-confidence fragment hits, which were subsequently validated using Affinity Selection Mass Spectrometry (ASMS) and Surface Plasmon Resonance (SPR) to determine binding affinities [26]. These case studies illustrate how semi-HTS enables the efficient management of library sizes that are substantial yet small enough to allow for immediate follow-up and validation.
Key Quantitative Data from Recent Helicase Screening Campaigns
The following table summarizes performance metrics from recent successful helicase inhibitor screening campaigns, highlighting the scale and efficiency of the semi-HTS approach.
Table 1: Performance Metrics of Recent Helicase Screening Campaigns
| Target | Screening Method | Library Size | Primary Hits | Confirmed Hits (IC50 <10 µM) | Key Assay Metrics |
|---|---|---|---|---|---|
| SARS-CoV-2 nsP13 [30] | Biochemical HTS (1,536-well) | ~650,000 compounds | 7,009 (1.08% hit rate) | 674 | Z' = 0.86 ± 0.05 |
| SARS-CoV-2 nsP13 [26] | Fragment-Based Drug Discovery (NMR) | ~500 fragments | 40 (8% hit rate) | N/A (KD determined) | Orthogonal confirmation via ASMS & SPR |
Essential Experimental Protocols
This section provides detailed methodologies for key experiments cited in the rationale for semi-HTS in helicase inhibitor discovery.
Semi-HTS Biochemical Assay for SARS-CoV-2 nsP13 Helicase
This protocol details the 1,536-well plate assay used to identify nsP13 inhibitor hit compounds [30].
- Principle: A double-stranded (ds)DNA substrate is labeled with a fluorophore (FAM) on one strand and a quencher (BHQ) on the other. Helicase unwinding activity displaces the quencher strand, leading to an increase in fluorescence. A trap DNA strand is included to prevent reannealing.
- Materials:
- Purified SARS-CoV-2 nsP13: Full-length, His-tagged protein in buffer (100 mM NaCl, 10 mM HEPES pH 7.4, 1 mM DTT) [30].
- dsDNA Substrate: Annealed strands (T20D25BHQ and FAM-T0D25) at 100 nM final concentration.
- Trap DNA: Unlabeled DNA strand (sequence: TCTAATGTAGTATAGTAATCCGCTC) at 500 nM final concentration.
- Assay Buffer: 100 mM NaCl, 2.5 mM MgCl2, 20 mM HEPES (pH 7.4), 2 mM ATP, 0.05% BSA.
- Stop Solution: 5X concentration (20 mM HEPES pH 7.4, 0.2 M NaCl, 0.2 M EDTA).
- Equipment: PHERAstar microplate reader (or equivalent), 1,536-well plates.
- Procedure:
- Dispense Enzyme/Trap Mixture: Pipette 2.5 µL of a mixture containing nsP13 (0.075 nM final concentration) and trap DNA in assay buffer into each well. For high-control wells (100% inhibition), dispense only trap DNA in assay buffer.
- Compound Addition: Transfer 30 nL of compound or DMSO control to the assay plate.
- Negative Control Setup: For negative control wells (0% inhibition), add 1 µL of 5X stop solution before proceeding to the next step.
- Initiate Reaction: Add 2.5 µL of dsDNA substrate (100 nM final concentration) to all wells.
- Centrifugation and Incubation: Centrifuge plates at 1,200 rpm for 1 minute to mix. Incubate at 30 °C for 30 minutes.
- Stop Reaction: Add 1 µL of 5X stop solution to all wells except the pre-stopped negative controls.
- Signal Detection: Measure the fluorescence intensity using a plate reader with excitation/emission filters of 485/520 nm.
- Data Analysis: Calculate percentage inhibition using the formula:
% Inhibition = (1 - (Signal_Compound - Signal_High Control) / (Signal_Low Control - Signal_High Control)) * 100. Z' factor is calculated to validate assay robustness.
Orthogonal Hit Confirmation Assays
The following orthogonal methods are critical for confirming specific helicase inhibition and mitigating false positives in a semi-HTS funnel [26].
- Fragment Screening by NMR:
- Protein Production: Express and purify NSP13 to ≥90% purity (yield ~0.5 mg/L) in PBS buffer (pH 7.4) with 100 µM DTT [26].
- Ligand-Observed NMR Experiments: Screen fragment cocktails using Saturation Transfer Difference (STD), WaterLOGSY, and T2 relaxation experiments. A positive binding event is indicated by an increase in STD signal, a change in WaterLOGSY signal, or an increase in ligand relaxation rates (T2/T1ρ) [26].
- Hit Confirmation: Identify "high-confidence" fragments that show positive responses and further characterize them using Diffusion-Ordered Spectroscopy (DOSY) to estimate binding affinity (KD).
- Affinity Selection Mass Spectrometry (ASMS):
- Procedure: Incubate compounds with NSP13 helicase at a fixed concentration. Separate the protein-ligand complexes from unbound compounds using size exclusion chromatography. Denature the complexes and identify bound ligands via mass spectrometry.
- Data Analysis: Compounds with a response ratio greater than three are selected as binders. Dose-response experiments are performed for selected hits to determine KD values [26].
- Surface Plasmon Resonance (SPR):
- Immobilization: Immobilize NSP13 protein on a sensor chip.
- Binding Analysis: Test analyte compounds (e.g., AMP-NP as a positive control, followed by hits from other screens) in a dose-dependent manner to measure binding kinetics and affinity (KD) [26].
Visualizing Screening Strategies and Pathways
Semi-HTS Screening Funnel for Helicase Inhibitors
The following diagram illustrates the multi-stage funnel used to identify and validate helicase inhibitors, from primary screening to confirmed hits.
Fragment-Based Discovery Workflow
This workflow outlines the fragment-based drug discovery process for identifying helicase inhibitors, as demonstrated for SARS-CoV-2 NSP13.
The Scientist's Toolkit: Research Reagent Solutions
The following table catalogues essential materials and reagents for implementing a semi-HTS campaign for helicase inhibitor discovery, as derived from the cited protocols.
Table 2: Essential Research Reagents for Helicase Semi-HTS
| Reagent / Material | Function in Assay | Example Specifications / Notes |
|---|---|---|
| Purified Helicase | Enzyme target for the screening assay. | Full-length SARS-CoV-2 nsP13 (aa 1-601) with His-tag; buffer: 100 mM NaCl, 10 mM HEPES pH 7.4, 1 mM DTT [30]. |
| Fluorescent Nucleic Acid Substrate | Report on helicase unwinding activity. | dsDNA with FAM fluorophore on one strand and BHQ quencher on the other; final concentration of 100 nM in assay [30]. |
| Trap Oligonucleotide | Prevents re-annealing of unwound strands to ensure signal stability. | Unlabeled DNA strand with sequence complementary to the displaced strand; used at 500 nM [30]. |
| ATP | Provides energy for the helicase's enzymatic activity. | Used at 2 mM in assay buffer as a cofactor [30]. |
| Fragment Library | Low molecular weight compounds for FBDD. | A curated collection of ~500 fragments for initial screening; allows identification of efficient binding motifs [26]. |
| SPR Chip | Immobilizes the protein for direct binding affinity and kinetics measurements. | Used in SPR assays to confirm binding of hits from primary screens and determine KD values [26]. |
Assay Formats and Implementation: Building Your Semi-HTS Workflow
Within the realm of molecular biology and drug discovery, helicases have emerged as critical therapeutic targets due to their indispensable roles in nucleic acid metabolism. The evaluation of helicase activity primarily hinges on two fundamental biochemical outputs: the measurement of ATP hydrolysis (ATPase activity) and the direct observation of nucleic acid strand separation (unwinding activity). For researchers engaged in semi-high-throughput screening (HTS), selecting the appropriate readout is paramount for campaign success. This application note provides a detailed comparative analysis of these core assay technologies, underpinned by specific experimental protocols and quantitative data to guide assay selection and implementation within a rigorous research framework.
Core Technology Principles and Comparison
Helicases are motor proteins that convert the chemical energy from nucleoside triphosphate hydrolysis (typically ATP) into mechanical work for unwinding double-stranded DNA or RNA. The two primary assay types monitor different stages of this catalytic cycle.
- ATPase Assays: These are indirect, universal assays that quantify the ADP produced from ATP hydrolysis [9] [31]. They report on the motor function of the helicase but do not directly confirm strand displacement.
- Unwinding Assays: These are direct, mechanistic assays that measure the physical separation of a double-stranded nucleic acid substrate into single strands, providing direct evidence of helicase function [32] [31].
The following diagram illustrates the fundamental workflows and decision-making process for selecting and implementing these core assay technologies.
Figure 1: A Sequential Assay Workflow for Helicase Screening
The table below summarizes the fundamental characteristics of these two assay approaches to facilitate direct comparison.
Table 1: Core Characteristics of Helicase Assay Technologies
| Feature | ATPase Assays | Unwinding Assays |
|---|---|---|
| Readout Principle | Quantification of ADP production [9] | Direct detection of strand separation [32] |
| Relationship to Function | Indirect measure of motor activity | Direct measure of biological function |
| Throughput | Very High (384-/1536-well) [30] [31] | High (384-well) [30] |
| Key Advantage | Universal; one assay for many helicases [31] | Mechanistically direct [31] |
| Key Limitation | Does not confirm unwinding; false positives from ATPase-only inhibitors [31] | Substrate-specific; potentially more complex [31] |
| Best Use Case | Primary HTS and inhibitor dose-response [31] | Hit validation, mechanistic studies, and substrate specificity [31] |
Quantitative Performance Data
The practical utility of an assay for screening is determined by its robustness and sensitivity. The following table collates quantitative performance metrics from published and commercial applications of both assay formats.
Table 2: Quantitative Assay Performance Metrics
| Assay Format / Target | Reported Z' Factor | Dynamic Range / Signal Change | Key Experimental Parameters |
|---|---|---|---|
| ATPase Assay (WRN Helicase) | 0.85 [33] | Linear ADP formation over 60 min [33] | 50 µM ATP, 40 nM DNA, 1 mM MgCl₂, 60 min @ 30°C [33] |
| ATPase Assay (SARS-CoV-2 nsP13) | ~0.86 (in HTS) [30] | Not Specified | 2 mM ATP, 2.5 mM MgCl₂, 30 min @ 30°C [30] |
| FRET Unwinding (SARS-CoV-2 nsP13) | Not Specified | 95% dsDNA unwound vs. 48% dsRNA [32] | 3 mM ATP, 5 mM MgCl₂ [32] |
| Fluorescent Dye Displacement | Not Specified | ~2x signal change [31] | Varies by substrate design |
The Z' factor is a statistical measure of assay robustness and quality, with values above 0.5 indicating excellent assays suitable for HTS. The data above demonstrate that both ATPase and unwinding assays can be optimized to meet this stringent requirement.
Detailed Experimental Protocols
Protocol 1: ATPase Activity Assay using ADP-Glo Technology
This protocol is adapted for a 384-well plate format and is ideal for semi-high-throughput profiling of helicase inhibitors [9] [30].
Research Reagent Solutions:
- Purified Helicase Enzyme: Recombinant protein (e.g., SARS-CoV-2 nsP13, WRN) [30] [33].
- DNA Substrate: A forked or tailed duplex DNA that stimulates ATPase activity (e.g., 37-bp 3'-Flap duplex for WRN) [4] [33].
- ATP Solution: Prepared in nuclease-free water [4].
- Assay Buffer: Typically contains Tris/HEPES (pH 7.4-7.5), NaCl, MgCl₂, and a non-ionic detergent [30] [33].
- ADP-Glo Max Reagents: Includes ADP-Glo Max Assay Kit (Promega, V7001) for high ATP concentration ranges [9].
Procedure:
- Reaction Setup: In a 384-well plate, dispense a 10 µL mixture containing:
- Incubation: Centrifuge the plate and incubate at 30°C for 30-60 minutes to allow the enzymatic reaction to proceed [30].
- ADP Detection: Add an equal volume of ADP-Glo Reagent to terminate the reaction and deplete remaining ATP. Incubate for 40-60 minutes at room temperature.
- Signal Development: Add the Kinase Detection Reagent to convert ADP to ATP and generate a luminescent signal. Incubate for 30-60 minutes at room temperature.
- Readout: Measure luminescence on a compatible plate reader. The signal is inversely proportional to helicase activity.
Protocol 2: FRET-Based Strand Displacement Unwinding Assay
This protocol uses a fluorescence resonance energy transfer (FRET) pair to monitor strand separation in real-time or at endpoint [32] [30].
Research Reagent Solutions:
- FRET-Labeled Duplex Substrate: A partial duplex nucleic acid with a fluorophore (e.g., FAM) on one strand and a quencher (e.g., BHQ) on the complementary strand [30].
- Trap DNA: An unlabeled oligonucleotide identical to the quencher-labeled strand, added in excess to prevent reannealing after unwinding [32] [30].
- Stop Solution: Contains EDTA (e.g., 20-40 mM) to chelate Mg²⁺ and terminate the reaction [30].
Procedure:
- Pre-incubation: In a low-volume 384-well plate, dispense a 2.5 µL mixture containing:
- Reaction Initiation: Initiate the unwinding reaction by adding 2.5 µL of the FRET-labeled dsDNA substrate (e.g., 100 nM final concentration) pre-mixed with ATP (e.g., 2 mM final) [30].
- Incubation: Centrifuge the plate and incubate at 30°C for 30-60 minutes.
- Reaction Termination: Add 1 µL of 5X Stop Solution (e.g., containing 200 mM EDTA) to quench the reaction [30].
- Readout: Measure the fluorescence intensity (Ex/Em: 485/520 nm for FAM) [30]. An increase in fluorescence indicates strand displacement and unwinding activity.
The following diagram contrasts the fundamental biochemical steps and detection methods for these two core protocols.
Figure 2: Biochemical Pathways for ATPase and Unwinding Assays
For a comprehensive helicase screening campaign, an integrated, sequential approach is highly recommended, leveraging the strengths of both technologies while mitigating their individual limitations.
The optimal strategy consists of a two-phase workflow:
- Primary HTS: Employ a robust, universal ATPase assay (e.g., Transcreener ADP² or ADP-Glo) to screen large compound libraries efficiently. This step identifies all compounds that interfere with the helicase's ATP hydrolysis motor [31].
- Orthogonal Hit Validation: Subject the confirmed hits from the primary screen to a direct unwinding assay (e.g., FRET-based displacement). This critical step filters out compounds that inhibit ATPase activity without affecting unwinding (false positives) and confirms true functional inhibitors that block the biological endpoint of strand separation [31].
This synergistic protocol ensures that only mechanistically validated hits progress to costly and time-consuming secondary assays and lead optimization, thereby de-risking the entire drug discovery pipeline. The quantitative data and detailed methodologies provided herein serve as a foundational guide for establishing a semi-high-throughput screening platform for helicase-targeted therapeutic development.
Helicases are essential molecular motors that unwind nucleic acids using the energy from ATP hydrolysis, playing critical roles in DNA replication, repair, recombination, transcription, and RNA metabolism [34]. Their therapeutic relevance is significant, with mutations in helicases like BLM and WRN linked to genomic instability in cancers, while RNA helicases such as RIG-I and DDX3 are vital for antiviral innate immune responses [34]. The discovery of inhibitors and modulators for these enzymes requires robust, sensitive, and high-throughput compatible assays. Homogeneous "mix-and-read" assays have emerged as the preferred format for high-throughput screening (HTS), eliminating separation steps, reducing hands-on time, and increasing throughput [34] [35] [36].
This application note details the implementation of two principal mix-and-read assay technologies for helicase activity: the Transcreener ADP2 Assay, which detects ADP production as a universal measure of ATPase activity, and Fluorescent Dye Displacement Assays, which directly measure strand separation. We provide validated protocols, performance data, and practical guidance to enable researchers to establish these assays for semi-high-throughput helicase activity screening and inhibitor profiling.
Key Assay Technologies and Comparison
The selection of an appropriate assay format depends on the specific research goals, whether for primary HTS, hit validation, or mechanistic studies. The table below summarizes the core characteristics of the two featured technologies.
Table 1: Comparison of Key Mix-and-Read Helicase Assay Technologies
| Feature | Transcreener ADP2 Assay | Fluorescent Dye Displacement (Heliscreener-type) |
|---|---|---|
| Principle | Immunoassay detecting ADP produced from ATP hydrolysis [37] | Direct measurement of fluorescence decrease as intercalating dye is displaced during DNA/RNA unwinding [34] |
| Target Activity | Indirect (ATPase activity) [34] | Direct (Unwinding activity) [34] |
| Throughput | High (384-/1536-well) [34] [37] | High (384-/1536-well) [34] |
| Key Reagents | ADP-Specific Antibody, Fluorescent ADP Tracer [37] | Fluorescently-labeled DNA/RNA substrate, Intercalating Dye |
| Z' Factor | ≥ 0.7 [34] [37] | ≥ 0.7 [34] |
| Best Use Cases | Primary HTS, Universal ATPase screening [34] | Orthogonal unwinding confirmation, Kinetic studies [34] |
The Scientist's Toolkit: Essential Research Reagent Solutions
The following table catalogues key materials required to establish these helicase activity assays.
Table 2: Essential Research Reagent Solutions for Helicase Assays
| Item | Function/Description | Example Application/Note |
|---|---|---|
| Transcreener ADP2 Assay Kit | Provides antibody, tracer, and buffers for a competitive FP, FI, or TR-FRET immunoassay to detect ADP [37]. | Universal for any ADP-generating enzyme; available in multiple readout configurations (FP, FI, TR-FRET) to match plate reader capabilities [38]. |
| Recombinant Helicase Enzyme | Active, purified helicase (e.g., WRN, BLM, DDX3). | Enzymes should be validated for activity; BellBrook offers active human WRN helicase with >90% purity [38]. |
| DNA/RNA Substrate | Optimized nucleic acid duplex for unwinding. | The Enzolution WRN Assay System includes a specific 40 μM WRN Helicase DNA substrate [38]. |
| White or Black Assay Plates | Low-volume, multi-well plates for HTS. | Use white plates for TR-FRET, black plates for FP/FI readouts [38]. |
| Multi-Mode Microplate Reader | Instrument capable of measuring fluorescence polarization (FP), intensity (FI), or time-resolved FRET (TR-FRET). | BMG LABTECH CLARIOstar and PHERAstar FS are certified for the Transcreener assay [37]. |
Experimental Protocols
Protocol 1: Helicase Activity and Inhibition Screening Using Transcreener ADP2 Assay
This protocol measures the ATPase activity of helicases like WRN by quantifying ADP production in a 384-well format, ideal for high-throughput inhibitor screening [38].
Materials and Reagents
- Enzolution WRN Helicase ATPase Assay System (includes recombinant WRN helicase, DNA substrate, and buffer) [38]
- Transcreener ADP2 Assay Kit (Choose FP, FI, or TR-FRET configuration) [38]
- Low-volume 384-well microplates (white for TR-FRET, black for FP/FI) [38]
- ATP and ultrapure nuclease-free water
- Test compounds (inhibitors) and DMSO
- Precision liquid handling equipment
- Compatible multi-mode microplate reader (e.g., CLARIOstar)
Procedure
- Plate Preparation: Dispense 1 μL of compound (in DMSO) or DMSO control into designated wells of a 384-well assay plate.
- Enzyme Reaction Mixture: Prepare the following mixture on ice:
- 1X Enzyme Assay Buffer A
- 1 mM MgCl₂
- 0.01% Triton X-100
- 40 nM WRN Helicase DNA substrate
- 50 μM ATP
- 0.20 – 0.65 nM WRN Helicase Enzyme
- Initiate Reaction: Add 9 μL of the enzyme reaction mixture to each well containing compound or DMSO. Seal the plate and incubate at 30°C for 60 minutes [38].
- Stop Reaction and Detect ADP: Prepare the ADP Detection Mixture appropriate for your readout. For the FP configuration, this contains 4 nM ADP2 AlexaFluor 633 Tracer and 55 μg/mL ADP2 Antibody in 1X Stop & Detect Buffer [38]. Add 10 μL of this Detection Mixture to each well. Seal the plate, incubate for 60 minutes at room temperature, and read the fluorescence polarization.
Data Analysis
- Generate an ADP standard curve by titrating ADP into ATP (e.g., 0-50 μM ADP in 50 μM total nucleotide) to convert raw mP values to ADP formed [38].
- Calculate percentage inhibition using positive (no enzyme) and negative (DMSO control) controls.
- For robust HTS, a Z' factor ≥ 0.7 is routinely achieved, indicating an excellent assay [38].
Diagram 1: Transcreener ADP2 assay workflow.
Protocol 2: Direct Unwinding Measurement via Fluorescent Dye Displacement
This protocol directly monitors the strand separation activity of helicases in real-time, providing orthogonal confirmation to ATPase assays [34].
Materials and Reagents
- Double-stranded DNA or RNA substrate (with a 5' or 3' overhang suitable for the target helicase)
- Fluorescent intercalating dye (e.g., SYBR Green) or pre-labeled molecular beacon-style substrates
- Assay buffer (typically containing Tris-HCl, MgCl₂, NaCl, and DTT)
- Active, purified helicase enzyme
- ATP
- 384-well optical bottom plates
Procedure
- Substrate Preparation: Generate a duplex nucleic acid substrate by annealing complementary strands. For a dye displacement assay, incubate the duplex with an intercalating dye. For a beacon-style assay (like Heliscreener), use a substrate where strand separation directly alters fluorescence [34].
- Plate Setup: In a 384-well plate, mix the following:
- Assay Buffer
- 1-50 nM fluorescently-labeled DNA/RNA substrate
- 1 mM ATP
- Test compound or control.
- Baseline Reading: Measure the baseline fluorescence for 5-10 minutes using a plate reader equipped with temperature control.
- Initiate Unwinding: Start the reaction by adding helicase enzyme to a final concentration determined by empirical titration (e.g., 0.1-10 nM).
- Kinetic Measurement: Immediately continue reading fluorescence continuously or at short intervals for 30-120 minutes. For dye displacement, a decrease in fluorescence is observed as the dye is released from the duplex. For some beacon assays, unwinding may cause an increase in fluorescence [34].
Data Analysis
- Normalize fluorescence signals to a no-enzyme control (0% unwinding) and a fully denatured substrate (100% unwinding).
- Plot normalized fluorescence vs. time to determine unwinding kinetics.
- For inhibitor screening, calculate % inhibition based on the initial rate of fluorescence change or the endpoint signal compared to controls.
Diagram 2: Fluorescent dye displacement workflow.
Results and Data Interpretation
Expected Outcomes and Performance Metrics
Both assays, when optimized, yield high-quality data suitable for semi-high-throughput screening.
- Robustness: The Transcreener ADP2 assay for WRN helicase consistently yields Z' factors ≥ 0.85, indicating an excellent and robust assay for HTS [38].
- Linearity: The ADP detection assay shows a linear response with time and enzyme concentration under initial velocity conditions, allowing for accurate kinetic analysis [38].
- Sensitivity: Pilot screens of a 1280-compound library using the Transcreener platform successfully identified inhibitors, with dose-response profiling yielding definitive IC₅₀ values (e.g., 30 nM for a potent hit) [38].
- Orthogonal Confirmation: A two-step workflow is highly recommended: using the universal ADP assay for primary HTS, followed by the direct unwinding assay for hit validation to confirm true enzymatic inhibition and rule out false positives that merely affect ATPase activity [34].
The implementation of homogeneous mix-and-read assays, specifically the Transcreener ADP2 and Fluorescent Dye Displacement platforms, provides a powerful, streamlined approach for semi-high-throughput screening of helicase activity and inhibition. The Transcreener assay offers a universal, robust, and HTS-ready solution for primary screening based on ATPase activity. The fluorescent unwinding assays provide direct, mechanistic insight and are ideal for orthogonal confirmation. Together, they form a comprehensive toolkit that accelerates the discovery of novel helicase inhibitors for therapeutic applications in oncology, antiviral therapy, and beyond.
Assay Configuration for 384-Well and 1536-Well Plate Formats
Helicases are motor proteins that utilize adenosine tri-phosphate (ATP) hydrolysis to unwind duplex nucleic acids, playing essential roles in fundamental cellular processes such as DNA replication, repair, RNA transcription, and translation [39] [40]. The critical nature of these functions makes helicases attractive therapeutic targets for antiviral and anticancer drug discovery [39]. High-throughput screening (HTS) serves as a core technology in modern drug discovery, enabling the rapid testing of thousands of chemical compounds to identify potential inhibitors or modulators of biological targets [41] [42]. The miniaturization of assays to 384-well and 1536-well plate formats represents a significant advancement, offering substantial economies in reagent consumption, cost, and time while dramatically increasing throughput capacity compared to conventional 96-well formats [43] [44]. This application note provides detailed methodologies and optimized parameters for configuring robust, miniaturized helicase activity assays suitable for semi-high-throughput screening campaigns, framed within the context of accelerating helicase-focused drug discovery research.
Helicase Assay Selection and Comparative Analysis
Selecting an appropriate assay format is the most critical step in developing a successful screening campaign. The ideal assay combines high sensitivity, robustness, throughput, and biological relevance while minimizing artifacts and false positives [39].
Major Helicase Assay Formats
The table below summarizes the principal biochemical assay formats available for detecting helicase enzyme activity, along with their key characteristics and recommended applications [39].
Table 1: Comparison of Major Helicase Assay Formats
| Format | Readout Principle | Advantages | Limitations | Best Use Cases |
|---|---|---|---|---|
| Gel-Based Unwinding | Separation of labeled duplex/unwound DNA or RNA via electrophoresis | Direct visualization of substrate and product; considered a gold standard for validation | Low throughput; labor-intensive and time-consuming | Mechanistic studies and orthogonal validation of primary hits |
| Fluorescent Dye Displacement | Decrease in fluorescence as an intercalating dye is released during unwinding | Continuous monitoring allows for kinetic studies; relatively simple setup | Dye may perturb the duplex structure; potential for compound interference | General kinetic studies and inhibitor characterization |
| Molecular Beacon/Hairpin | Fluorescence change (increase or decrease) upon hairpin opening/strand separation | Real-time, customizable substrate design | Requires careful optimization of substrate design complexity | Mid-to-high-throughput kinetic assays and screening |
| ADP Detection (e.g., Transcreener ADP²) | Detects ADP produced as a universal product of ATP hydrolysis | Homogeneous, "mix-and-read" format; universal for any ATP-dependent enzyme; HTS-ready | Indirectly measures ATPase activity, not unwinding directly | Primary HTS, broad applicability across helicase families |
| Unwinding (e.g., Heliscreener) | Fluorescence increases as helicase separates fluorophore-quencher labeled strands | Directly measures unwinding; real-time, high-throughput, and sensitive | Requires an optimized and sometimes costly substrate | Primary HTS, kinetics, and definitive inhibitor profiling |
For most drug discovery workflows, fluorescence-based strand displacement assays (like the Heliscreener platform) and homogeneous ADP detection assays (like the Transcreener ADP² platform) offer the optimal balance of sensitivity, robustness, and throughput. These "mix-and-read" assays typically deliver high Z′ factors (≥ 0.7) and low false-positive rates, making them ideal for HTS of helicase inhibitors or modulators [39].
Assay Selection Workflow
The following diagram outlines a logical decision-making workflow for selecting the most appropriate helicase assay based on the screening goals and resources.
Experimental Protocols
This section provides detailed, step-by-step protocols for implementing helicase activity assays in both 384-well and 1536-well microplate formats.
Molecular Beacon-Based Helicase Assay in 384-Well Format
This protocol describes a fluorescence-based method to monitor helicase-catalyzed DNA unwinding in real-time, adapted for a 384-well plate [40].
Materials & Reagents
- White, 384-well, low-volume microplates (e.g., Corning)
- Microplate reader equipped with injectors and capable of kinetic fluorescence measurements (e.g., BMG LABTECH)
- Assay Buffer: 25 mM MOPS, pH 6.5, 1.25 mM MgCl₂, 5 μg/mL BSA, 0.001% (v/v) Tween-20, 50 μM DTT
- Molecular Beacon Substrate: Double-stranded DNA with one strand carrying a fluorophore (e.g., Cy5) and a quencher (e.g., IAbRQ) [40].
- ATP Solution: 10 mM in assay buffer (for injection)
- Helicase Enzyme: Purified recombinant protein (e.g., HCV NS3 helicase)
- Test Compounds: Dissolved in DMSO
Procedure
- Substrate Preparation: Anneal the DNA strands to create the molecular beacon substrate. Prepare a working stock of 5-10 nM in assay buffer.
- Reaction Mix Preparation: Prepare a master mix containing assay buffer, molecular beacon substrate (e.g., 5 nM final), BSA, DTT, and helicase enzyme (e.g., 12.5 nM final). Gently mix and avoid introducing bubbles.
- Plate Seeding: Dispense the reaction mix into the 384-well plate at a volume of 35 μL per well [43].
- Compound Addition: Add test compounds to designated wells. Include controls (e.g., negative control with DMSO only; positive control with a known inhibitor like 100 μM primuline). The final DMSO concentration should be normalized (e.g., 1-5%) across all wells.
- Initial Reading: Place the plate in the pre-warmed microplate reader. Read fluorescence for 2-5 minutes (Cycle 1) to establish a stable baseline. Instrument Settings: Fluorescence intensity, top optics; Filters: Ex/Em 640-10/680-10 for Cy5; Cycle time: 5-20 seconds [40].
- Reaction Initiation: After the baseline reading, inject ATP into all wells from the injector to a final concentration of 1 mM. Use smart dispensing if available to minimize timing differences between wells.
- Kinetic Measurement: Continue reading fluorescence immediately after injection for 30-60 minutes to monitor the decrease in signal as the substrate is unwound.
- Data Analysis: Using the plate reader's software (e.g., MARS Data Analysis Software):
- Identify the linear range of the signal decrease after ATP injection.
- Calculate the slope of the fluorescence decrease for each well, which corresponds to the initial rate of unwinding.
- For inhibitor screening, plot the slope (as % activity) against the log of inhibitor concentration and fit a 4-parameter curve to determine the IC₅₀ value.
Homogeneous ADP Detection Assay in 1536-Well Format
This protocol uses a universal, homogenous ADP detection method to screen for helicase inhibitors based on their ATPase activity, miniaturized for a 1536-well plate [43] [39].
Materials & Reagents
- White, 1536-well microplates (tissue culture treated)
- ADP Detection Reagent: Homogeneous immunoassay-based kit (e.g., Transcreener ADP² FRET or BellBrook Labs' platform)
- Assay Buffer: Optimized for the specific helicase target (typically containing Tris or HEPES, Mg²⁺, NaCl)
- ATP Solution: Prepared in assay buffer
- DNA/RNA Substrate: Unlabeled duplex nucleic acid relevant to the helicase being tested.
- Helicase Enzyme: Purified recombinant protein.
- Test Compounds: In DMSO.
Procedure
- Plate Preparation: Using an automated liquid handler (e.g., Perkin-Elmer Janus with a 384-pin head), transfer 1 μL of each test compound in DMSO to the 1536-well plate [45].
- Reaction Mix Preparation: Prepare a master mix containing assay buffer, helicase enzyme, and nucleic acid substrate. Keep on ice.
- Dispensing Reaction Mix: Dispense 5 μL of the reaction mix into each well of the 1536-well plate using a precision dispenser (e.g., BioTek Multiflo with a 1 μL cassette) [43]. Centrifuge the plate briefly at 1,000 RPM for 1 minute to ensure all liquid is at the bottom of the wells [43].
- Pre-incubation: Seal the plate and pre-incubate for 15-30 minutes at room temperature to allow compounds to interact with the enzyme.
- Reaction Initiation: Using the plate reader's injector, add 2 μL of ATP solution to each well to start the reaction. The final total assay volume will be 8 μL [43] [45]. The final ATP concentration must be determined during assay optimization (often in the micromolar range).
- Incubation and Detection: Incubate the plate for a predetermined time (e.g., 30-60 minutes at room temperature or 37°C). Then, add the homogeneous ADP detection reagent according to the manufacturer's instructions. This often involves a single addition step with no washing required.
- Endpoint Reading: Read the plate using a multilabel plate reader (e.g., Perkin-Elmer Wallac Envision) configured for FRET or fluorescence polarization, as required by the detection kit.
- Data Analysis:
- Calculate the % inhibition for each compound using positive (no enzyme or high-dose inhibitor) and negative (DMSO control) controls on the same plate.
- The Z' factor, a measure of assay robustness, should be ≥ 0.7 for a high-quality HTS assay [39] [45]. It is calculated as: Z' = 1 - (3σc+ + 3σc-) / |μc+ - μc-|, where σ and μ are the standard deviations and means of the positive (c+) and negative (c-) controls [45].
Experimental Workflow
The diagram below illustrates the integrated experimental workflow for a high-throughput screening campaign, from plate preparation to data analysis.
The Scientist's Toolkit: Research Reagent Solutions
Successful implementation of miniaturized assays depends on the use of specific, high-quality reagents and instruments. The following table details essential materials and their functions.
Table 2: Essential Research Reagents and Equipment for Miniaturized Helicase Assays
| Item | Function / Role in the Assay | Example Vendor / Specification |
|---|---|---|
| gWiz-Luc/gWiz-GFP Plasmid | Reporter plasmid for transfection control or cell-based assays; contains luciferase or GFP gene under CMV promoter. | Aldevron [43] |
| Polyethylenimine (PEI) | Cationic polymer used for forming polyplexes with DNA for transient transfection in cell-based systems. | Sigma-Aldrich (25 KDa) [43] |
| ONE-Glo Luciferase Assay | Luciferase detection reagent for endpoint bioluminescence reading in cell-based reporter assays. | Promega [43] |
| Transcreener ADP² Assay | Homogeneous, HTS-ready immunoassay for detecting ADP produced by ATP-consuming enzymes like helicases. | BellBrook Labs [39] |
| Alamar Blue (Resazurin) | Cell-permeant redox indicator; fluorescence increases with metabolic activity, used in cell viability assays. | Serotec Ltd [45] |
| M13mp18 Single-Strand DNA | Common template for preparing double-stranded DNA substrates for helicase unwinding assays. | New England Biolab (N4040S) [46] |
| IgG Sepharose 6 Fast Flow | Resin for immobilizing tagged proteins (e.g., IgG-IgG-TEV tagged helicases) for pull-down or activity assays. | GE Healthcare [46] |
| Automated Liquid Handler | For precise, high-speed transfer of compounds and reagents in 384/1536-well formats (e.g., 384-pin head). | Perkin-Elmer Janus [43] |
| Multidrop Combi / FlexDrop | Reagent dispenser for rapid, uniform addition of cell suspensions and reagents to 384/1536-well plates. | Perkin Elmer [45] [47] |
| Multilabel Plate Reader | Detects absorbance, fluorescence, luminescence; equipped with injectors for kinetic assays (e.g., Envision). | Perkin-Elmer Wallac Envision [43] |
The successful configuration of helicase activity assays in 384-well and 1536-well formats provides a powerful platform for semi-high-throughput and ultra-high-throughput screening in drug discovery research. As demonstrated, the miniaturization of these assays to total volumes of 35 μL and 8 μL, respectively, is not only feasible but also results in robust performance, as indicated by Z' factors exceeding 0.5 and often reaching 0.7-0.9 in optimized systems [43] [45]. The choice between a direct unwinding assay (like the molecular beacon format) and an indirect ATPase assay (like ADP detection) depends on the specific research goals, available resources, and the need for direct mechanistic confirmation. By adhering to the detailed protocols, optimization parameters, and reagent specifications outlined in this application note, researchers can reliably establish these miniaturized formats in their own laboratories. This will significantly accelerate the identification and characterization of novel helicase inhibitors, ultimately contributing to the development of new therapeutic agents for cancer, viral infections, and other diseases.
Helicases are essential motor proteins that unwind nucleic acid duplexes, playing critical roles in genome replication, repair, and transcription. Their dysregulation is implicated in viral replication and cancer, making them attractive therapeutic targets. This application note details semi-high-throughput screening (HTS) campaigns targeting two therapeutically significant helicases: SARS-CoV-2 Non-Structural Protein 13 (Nsp13) and Human Werner Syndrome Helicase (WRN).
The SARS-CoV-2 Nsp13 helicase is indispensable for viral replication and is highly conserved among coronaviruses, presenting an ideal target for broad-spectrum antiviral therapeutics [48] [49]. The human WRN helicase, critical for DNA repair and genomic stability, has emerged as a promising synthetic lethal target for treating microsatellite instability-high (MSI-H) cancers [50] [22]. This case study outlines the development, optimization, and implementation of biochemical and cell-based assays to identify and characterize novel inhibitors of these helicases, providing a framework for future helicase-targeted drug discovery efforts.
Table 1: Key Characteristics of Nsp13 and WRN Helicases
| Feature | SARS-CoV-2 Nsp13 | Human WRN |
|---|---|---|
| Primary Function | Viral RNA replication, 5' mRNA capping, RNA unwinding [49] | DNA repair, replication, telomere maintenance, genomic stability [51] [22] |
| Enzymatic Activities | dsDNA/RNA unwinding, NTP hydrolysis, RNA 5'-triphosphatase [49] [17] | DNA unwinding, 3'→5' exonuclease, ATP hydrolysis [51] [22] |
| Directionality | 5' to 3' unwinding [49] | 3' to 5' unwinding [51] |
| Therapeutic Rationale | Essential, highly conserved viral enzyme; inhibition blocks replication [48] [49] [17] | Synthetic lethality in MSI-H cancers; essential for MSI-H but not MSS cell survival [50] [52] [22] |
| Disease Relevance | COVID-19 and coronaviridae infections [49] [17] | MSI-H cancers (colorectal, endometrial, gastric), Werner syndrome [50] [22] |
Quantitative Assay Parameters and Screening Outcomes
Table 2: Summary of HTS Campaign Parameters and Results
| Parameter | SARS-CoV-2 Nsp13 HTS [17] | WRN Biochemical Screening [51] | WRN Cell-Based Screening [50] |
|---|---|---|---|
| Screening Format | 1,536-well plate | 96-well (semi-HTS) | 384-well plate |
| Library Size | ~650,000 compounds | 500 compounds (NCI Diversity Set) | Horizon OncoSignature panel (301 cell lines) |
| Primary Assay | Biochemical helicase activity | Radiometric helicase assay | Cell viability/proliferation |
| Z'-Factor | 0.86 ± 0.05 | Not specified | Not specified |
| Primary Hit Rate | 1.08% (7,009 compounds) | 1.4% (7 compounds) | MSI-H selective inhibition |
| Confirmed Hits | 1,763 compounds | 2 lead compounds (NSC 19630, NSC 617145) | Multiple series including spirocyclic inhibitors |
| Potency Range (IC₅₀/GI₅₀) | IC₅₀ <10 μM (674 compounds) | NSC 19630: IC₅₀ ~20 μM; NSC 617145: more potent [51] | HRO761: GI₅₀ 50-1,000 nM (MSI-H cells) [22] |
Experimental Protocols
SARS-CoV-2 Nsp13 Biochemical HTS Protocol
Objective: Identify inhibitors of Nsp13 helicase activity in a 1,536-well plate format [17].
Materials:
- Recombinant SARS-CoV-2 Nsp13 protein
- Double-stranded DNA or RNA substrate with fluorescent labeling
- Reaction buffer: 25 mM HEPES (pH 7.5), 5 mM MgCl₂, 1 mM DTT, 0.01% Tween-20
- ATP solution (5 mM stock)
- Compound library (dissolved in DMSO)
- Stop solution: EDTA and detection reagents
Procedure:
- Assay Setup: Dispense 2 µL of reaction buffer into each well of the 1,536-well plate.
- Compound Addition: Transfer 10 nL of compound solutions (or DMSO control) via acoustic dispensing.
- Enzyme Addition: Add 2 µL of Nsp13 solution (final concentration 5-10 nM) to all wells.
- Reaction Initiation: Add 2 µL of substrate/ATP mixture (final concentrations: 50 nM substrate, 1 mM ATP).
- Incubation: Incubate plates at room temperature for 60 minutes.
- Reaction Termination: Add 2 µL stop solution containing EDTA and fluorescent detection reagents.
- Signal Detection: Measure fluorescence intensity using a plate reader with appropriate excitation/emission filters.
- Data Analysis: Calculate percentage inhibition relative to DMSO and compound-free controls.
Quality Control: Include controls in each plate: no enzyme (background), DMSO only (full activity), and reference inhibitor if available. Maintain DMSO concentration ≤1% [51] [17].
WRN Helicase Biochemical Screening Protocol
Objective: Screen compound libraries for WRN helicase inhibitors using a radiometric assay [51].
Materials:
- Purified recombinant WRN protein (devoid of nuclease contamination)
- ³²P- or fluorescently-labeled partial duplex DNA substrate
- Helicase reaction buffer: 30 mM HEPES (pH 7.8), 7 mM MgCl₂, 4 mM ATP, 40 mM creatine phosphate, 10 μg/mL creatine phosphokinase, 1 mM DTT, 100 μg/mL BSA
- Compound library (50 μM final concentration in screening)
- Non-denaturing polyacrylamide gel electrophoresis (PAGE) supplies
- Gel imaging system (phosphorimager or fluorescence scanner)
Procedure:
- Reaction Setup: Prepare master mix containing reaction buffer, DNA substrate (0.5 nM), and energy regeneration system.
- Compound Addition: Aliquot 19 μL master mix per reaction tube; add 1 μL compound solution or DMSO control.
- Enzyme Addition: Add WRN protein (concentration pre-titered to achieve 50-75% substrate unwound).
- Incubation: Incubate at 37°C for 15-30 minutes.
- Reaction Termination: Add 5 μL stop solution (50 mM EDTA, 0.5% SDS, 0.1% bromophenol blue, 40% glycerol) with 100-fold excess unlabeled oligonucleotide.
- Product Separation: Load samples on non-denaturing PAGE gel (6-8% acrylamide).
- Electrophoresis: Run at 200V for 1.5-2 hours in 0.5× TBE buffer.
- Visualization and Quantification: Expose gel to phosphorimager screen or scan fluorescence; quantify using ImageQuantTL or similar software.
- Data Analysis: Calculate percentage unwinding = (unwound product / total substrate) × 100 [51].
WRN Cellular Target Engagement Assay
Objective: Confirm functional inhibition of WRN in MSI-H cells by measuring DNA damage response [50] [22].
Materials:
- MSI-H and MSS cell lines (e.g., SW48, HCT116 for MSI-H; appropriate MSS controls)
- Test compounds and DMSO vehicle
- Antibodies: anti-phospho-H2AX (Ser139), anti-WRN, anti-p53, loading control
- Cell culture reagents and equipment
- High-content imaging system or Western blot supplies
Procedure:
- Cell Seeding: Plate MSI-H and MSS cells in 384-well plates at optimized density.
- Compound Treatment: Treat cells with test compounds (serially diluted) and controls for 72-96 hours.
- Fixation and Staining: Fix cells, permeabilize, and immunostain for pH2AX and other markers.
- Image Acquisition: Acquire images using high-content imaging system (20× objective).
- Image Analysis: Quantify nuclei count, pH2AX intensity, and other parameters.
- Orthogonal Validation: Perform Western blotting for WRN, pH2AX, p53, and chromatin-bound protein fractions.
- Data Interpretation: WRN inhibition specifically induces DNA damage response (pH2AX foci) in MSI-H but not MSS cells [50] [22].
Research Reagent Solutions
Table 3: Essential Research Reagents for Helicase HTS Campaigns
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Helicase Proteins | Recombinant SARS-CoV-2 Nsp13 [49] [17], Full-length WRN [51] [22] | Target enzymes for biochemical assays |
| Nucleic Acid Substrates | Fluorescently-labeled (Cy5, FAM) partial duplex DNA/RNA [17], ³²P-labeled DNA substrates [51] | Helicase activity measurement |
| Detection Technologies | ADP-Glo ATPase assay [50] [26], FRET-based unwinding assays [17] [26] | Enzyme activity readout |
| Cellular Models | MSI-H cell lines (SW48, HCT116) [50] [22], MSS isogenic controls [50] | Cellular target engagement and specificity testing |
| Key Assay Components | Non-denaturing PAGE gels [51], 1,536-well plates [17], high-content imaging systems [50] | Assay implementation and readout |
Experimental Workflows and Signaling Pathways
SARS-CoV-2 Nsp13 HTS Screening Funnel
WRN Inhibitor Screening and Validation Strategy
WRN Inhibition Mechanism in MSI-H Cells
This case study demonstrates successful implementation of semi-high-throughput screening strategies for two therapeutically significant helicases. The SARS-CoV-2 Nsp13 campaign employed a robust biochemical HTS approach, screening over 650,000 compounds to identify promising antiviral leads [17]. The WRN program integrated biochemical, biophysical, and extensive cell-based profiling to develop inhibitors that selectively target MSI-H cancers through synthetic lethality [50] [22].
Key success factors included the use of orthogonal assay formats for hit confirmation, incorporation of counter-screens to eliminate non-specific compounds, and careful attention to assay quality metrics. For WRN, the combination of ATP-binding assays with functional helicase assays proved crucial for identifying authentic allosteric inhibitors [22]. These case studies provide validated frameworks for future helicase-targeted drug discovery programs, highlighting both common principles and target-specific adaptations required for success.
Helicases are essential molecular motors that unwind nucleic acids using ATP hydrolysis, playing critical roles in DNA replication, repair, transcription, and RNA metabolism [53]. Their fundamental functions in maintaining genomic stability make them attractive targets for therapeutic intervention, particularly in oncology and antiviral therapy [53]. For instance, mutations in BLM and WRN helicases cause genomic instability and are linked to synthetic lethal vulnerabilities in cancer, while RNA helicases such as RIG-I and DDX3 are crucial for viral RNA detection and interferon signaling activation [53]. The complexity of helicase function—often within multi-protein complexes with intricate regulatory mechanisms—presents unique challenges for inhibitor discovery that require carefully designed screening strategies [54].
This application note provides a comprehensive framework for implementing a multi-stage screening cascade specifically tailored for helicase drug discovery. We outline robust experimental protocols, data analysis techniques, and decision-making gateways that progress from primary high-throughput screening (HTS) through rigorous hit validation, utilizing the latest technological advances in biochemical and cell-based assays to identify and characterize novel helicase inhibitors with high confidence.
Helicase Assay Selection and Development
Assay Format Comparison
Selecting appropriate assay formats is crucial for successful helicase inhibitor screening. The ideal assay combines high sensitivity, robustness, throughput, and biological relevance while minimizing artifacts. The table below summarizes the principal biochemical formats used to detect helicase enzyme activity:
Table 1: Comparison of Major Helicase Assay Formats
| Format | Readout Principle | Advantages | Limitations | Best Use Cases |
|---|---|---|---|---|
| Gel-based Unwinding | Separation of labeled duplex/unwound DNA or RNA | Direct visualization; gold standard | Low throughput; laborious | Mechanistic validation |
| Fluorescent Dye Displacement | Decrease in fluorescence as intercalating dye released during unwinding | Continuous, simple | Dye may perturb duplex; compound interference | General kinetic studies |
| Unwinding (Heliscreener) | Fluorescence increases as helicase separates strands with FRET or quencher pairs | Real-time, high-throughput, sensitive | Requires optimized substrate design | HTS, kinetics, inhibitor profiling |
| 2-Aminopurine Incorporation | Quenching loss as base unpairs | Label-minimal, simple | Low signal change (~2×) | Mechanistic follow-up |
| ADP Detection (Transcreener ADP²) | Detects ADP produced from ATP hydrolysis using immunodetection | Universal, homogeneous, HTS-ready | Indirect (measures ATPase activity only) | Primary screening, broad applicability |
| Molecular Beacon/Hairpin | Fluorescence change upon hairpin opening | Real-time, customizable | Substrate design complexity | Mid-throughput kinetic assays |
For high-throughput screening applications, homogeneous "mix-and-read" assays such as ADP detection platforms and fluorescence-based strand displacement assays typically offer the optimal balance of performance characteristics [53]. These formats deliver high Z′ factors (≥0.7) and low false-positive rates, making them ideal for primary screening of large compound libraries [53].
Key Research Reagent Solutions
Successful implementation of helicase screening assays requires access to well-characterized reagents and specialized detection systems. The following table outlines essential research tools and their applications in helicase drug discovery:
Table 2: Essential Research Reagent Solutions for Helicase Screening
| Reagent/System | Function/Application | Key Features |
|---|---|---|
| Transcreener ADP² Assay | Universal detection of ADP produced by helicase ATPase activity | Homogeneous, mix-and-read format; compatible with 384-/1536-well plates; Z′ factors ≥ 0.7 |
| Heliscreener Platform | Direct measurement of DNA/RNA unwinding via fluorescence | Real-time kinetic monitoring; high sensitivity; minimal compound interference |
| Enzolution WRN Helicase ATPase Assay System | Pre-optimized system for screening WRN helicase inhibitors | Includes purified human WRN helicase and DNA substrate; validated for HTS |
| Bst-DNA Polymerase | Essential for helicase-dependent amplification (HDA) applications | Strand-displacing activity; thermostable; used in isothermal amplification |
| Tte-UvrD Helicase | Thermophilic helicase for helicase-dependent amplification | Thermostable; unwinds DNA at 60-65°C; eliminates need for accessory proteins |
| Low-Volume Assay Plates | Microplate platform for HTS | Polystyrene non-binding surface; minimizes reagent consumption |
Multi-Stage Screening Cascade Workflow
Implementing a tiered screening cascade with multiple decision points is essential for efficiently progressing from initial screening to validated hits. The following workflow diagram illustrates the comprehensive multi-stage approach:
Primary Screening Phase
High-Throughput Screening Protocol
Objective: Identify initial hit compounds with inhibitory activity against target helicase from large compound libraries (10,000-100,000+ compounds).
Materials:
- Transcreener ADP² FP Assay Kit (BellBrook Labs) or equivalent
- Purified helicase enzyme (e.g., WRN, BLM, DDX3)
- Appropriate DNA/RNA substrate (forked DNA for DNA helicases)
- 384-well low volume assay plates (Corning #4514)
- Automated liquid handling system
- Multimode plate reader capable of FP, FI, or TR-FRET detection
Procedure:
- Reaction Setup: Prepare enzyme reaction mix containing:
- 1X Enzyme Assay Buffer (50 mM Tris pH 7.5, 1 mM MgCl₂, 0.01% Triton)
- Helicase enzyme (optimized concentration, typically 1-10 nM)
- DNA substrate (optimized concentration, typically 10-100 nM)
- ATP (Km concentration, typically 10-100 μM)
Compound Addition: Using automated liquid handler:
- Transfer 100 nL of compound solution (10 mM in DMSO) to assay plates
- Include controls: DMSO only (negative control), known inhibitor (positive control)
Reaction Initiation: Add 10 μL of enzyme reaction mix to each well
- Centrifuge plates briefly (1000 × g, 1 minute) to mix
Incubation: Incubate at optimal temperature (e.g., 30°C for WRN) for 30-60 minutes
Detection: Add 10 μL of Stop & Detect Buffer containing:
- 1X Stop & Detect Buffer (20 mM HEPES pH 7.5, 20 mM EDTA, 0.02% Brij-35)
- ADP2 Antibody (final concentration 30-60 nM)
- Alexa Fluor 633 Tracer (final concentration 40-80 nM)
Signal Measurement: Incubate 10-30 minutes, then read fluorescence polarization on compatible plate reader
Data Analysis:
- Calculate % inhibition = (1 - (Signalcompound - Signalpositive)/(Signalnegative - Signalpositive)) × 100
- Apply quality control metrics: Z′ factor ≥ 0.7, signal-to-background ratio ≥ 3
- Set hit threshold: Typically >50% inhibition or >3 standard deviations from mean
This primary screening approach successfully identified Mcm2-7 inhibitors with a 0.9% hit rate from a 1280-compound library screen [54].
Dose-Response Confirmation
Objective: Confirm activity of primary hits and determine preliminary potency (IC₅₀ values).
Procedure:
- Compound Dilution: Prepare 10-point, 2-fold serial dilutions in DMSO using automated liquid handling
- Plate Setup: Transfer diluted compounds to assay plates in triplicate
- Assay Execution: Follow primary screening protocol with dose-ranging compounds
- Data Analysis: Fit dose-response curves using four-parameter logistic equation:
- Y = Bottom + (Top - Bottom) / (1 + 10^((LogIC₅₀ - X) × HillSlope))
This confirmation step typically reduces hit rates from ~0.9% to ~0.4% [54].
Hit Validation Phase
Orthogonal Assay Implementation
Objective: Confirm hits using a different assay format to eliminate technology-specific artifacts.
Materials and Protocol: For helicase targets, implement a direct unwinding assay (e.g., Heliscreener) that measures strand separation rather than ATP hydrolysis:
- Substrate Design: Prepare forked DNA substrate with fluorophore-quencher pair
- Reaction Conditions: Similar to primary assay but monitoring fluorescence increase directly
- Compound Testing: Test confirmed hits from primary screen in dose-response format
- Data Analysis: Calculate IC₅₀ values and compare with primary assay results
Compounds showing consistent activity across both ATPase and unwinding assays progress to counter-screening.
Counter Assay and Selectivity Screening
Objective: Eliminate compounds with non-specific mechanisms of action and assess selectivity across related targets.
Procedure:
- Counter Assays: Test compounds against:
- Unrelated enzymes with similar cofactors (e.g., other ATPases)
- Assay components alone (detection systems, substrates)
Selectivity Panels: Profile compounds against:
- Related helicase family members (e.g., other RecQ helicases)
- Anti-targets associated with potential toxicity
Compound Interference Testing: Assess potential assay interference:
- Fluorescence quenching/interference
- Protein aggregation potential
- Redox activity
The following workflow diagram illustrates the key components of a comprehensive hit validation process:
Biomolecular Interaction Studies
Objective: Confirm direct binding to target helicase and determine binding affinity.
Protocol: Surface Plasmon Resonance (SPR) for Helicase-Ligand Interactions
- Immobilization: Covalently immobilize purified helicase on CMS sensor chip via amine coupling
- Binding Measurements: Inject compound dilutions (0.1-100 μM) in running buffer (HBS-EP+)
- Kinetic Analysis: Monitor association (60-120 seconds) and dissociation (120-300 seconds) phases
- Data Processing: Fit sensorgrams to 1:1 binding model to determine Kd, kon, and koff values
For the Mcm2-7 helicase, this approach confirmed direct binding of a validated hit (CMA) with Kdapp of 119 μM [54].
Hit Qualification Phase
Mechanistic and Functional Characterization
Objective: Elucidate mechanism of action and cellular activity of validated hits.
Protocol: Cell-Based Helicase Inhibition Assay
- Strain Engineering: Utilize isogenic cell pairs (wild-type vs. helicase mutant) for synthetic lethality screening [54]
- Growth Inhibition: Treat cells with compound dilutions (0-100 μM) for 24-48 hours
- Viability Assessment: Measure growth kinetics via absorbance (Abs600) every hour
- Data Analysis: Calculate differential growth inhibition using linear discriminant analysis (LDA)
Protocol: DNA Replication Inhibition Assessment
- Cell Synchronization: Synchronize cells in G1 phase using appropriate methods
- Compound Treatment: Add compounds at G1/S transition
- Cell Cycle Analysis: Fix cells at time intervals, stain DNA with propidium iodide
- FACS Analysis: Measure DNA content by flow cytometry to assess S-phase progression
This approach demonstrated that CMA specifically blocked S-phase progression in yeast cells at slightly higher concentrations than required for in vitro binding [54].
Initial SAR and Compound Profiling
Objective: Establish preliminary structure-activity relationships and assess drug-like properties.
Procedure:
- Analog Sourcing: Acquire or synthesize structural analogs of validated hits
- SAR Testing: Profile analogs in primary and orthogonal assays
- Property Assessment: Evaluate key physicochemical and ADME properties:
- Solubility (kinetic, thermodynamic)
- Metabolic stability (microsomal, hepatocyte)
- Membrane permeability (PAMPA, Caco-2)
- Plasma protein binding
- IP Assessment: Conduct patent landscape analysis
Data Analysis and Quality Control
Statistical Rigor and QC Metrics
Robust statistical analysis is essential throughout the screening cascade. Implement these key quality metrics:
Table 3: Essential Quality Control Metrics for Helicase Screening
| Stage | QC Metric | Target Value | Purpose |
|---|---|---|---|
| Primary Screening | Z′ factor | ≥ 0.7 | Assay robustness and suitability for HTS |
| Primary Screening | Signal-to-Background | ≥ 3:1 | Sufficient dynamic range |
| Primary Screening | Coefficient of Variation (CV) | < 10% | Well-to-well reproducibility |
| Hit Confirmation | R² of dose-response | > 0.9 | Quality of curve fitting |
| Hit Confirmation | Hill Slope | 0.5-2.0 | Appropriate binding characteristics |
| Orthogonal Assay | Correlation (r) with primary | > 0.7 | Consistency across assay formats |
For cell-based assays, multivariate analysis approaches such as Linear Discriminant Analysis (LDA) can consolidate multiple growth parameters into a single metric that fully describes compound effects on cell proliferation [54].
Data Management and Visualization
Effective data management strategies include:
- Compound tracking via barcode systems
- Automated data processing pipelines
- Activity cliffs visualization for SAR analysis
- Chemoinformatic analysis for scaffold diversity assessment
The multi-stage screening cascade outlined here provides a robust framework for identifying and validating helicase inhibitors with high confidence. By implementing orthogonal assay formats, rigorous counter-screening, and comprehensive mechanistic studies, researchers can efficiently progress from primary HTS to qualified hits with defined mechanism of action and promising pharmacological properties.
The field continues to evolve with emerging technologies such as cryo-EM for structural characterization of helicase-inhibitor complexes [55], single-molecule techniques for detailed mechanistic studies [3], and advanced cellular models for physiological relevance. Integrating these approaches into the screening cascade will further enhance our ability to develop targeted helicase inhibitors as novel therapeutic agents for cancer, viral infections, and other diseases.
Achieving Robust Performance: Troubleshooting and Critical Optimization Steps
In the realm of semi-high-throughput screening (HTS) for helicase activity, the reliability of an assay is paramount for successful drug discovery. For researchers aiming to identify novel helicase inhibitors or modulators, the consistency and quality of the primary screen directly impact the validity of downstream hits. Three statistical parameters serve as critical benchmarks for assay performance and data integrity: the Z'-factor for overall assay quality, the Signal-to-Background Ratio (S/B) for dynamic range, and the Coefficient of Variation (CV) for precision [56] [57] [58]. This application note details the interpretation and application of these metrics within the specific context of biochemical helicase activity assays, providing protocols and frameworks to ensure the generation of robust, reproducible data for research professionals.
Key Performance Metrics: Definitions and Interpretations
Z'-factor
The Z'-factor is a statistical parameter used exclusively during assay development and validation to assess the quality and suitability of an assay for high-throughput screening. It evaluates the separation between the positive and negative control signals and the data variation associated with these controls [56] [59]. The Z'-factor is defined by the equation:
[ Z'\text{-factor} = 1 - \frac{3(\sigmap + \sigman)}{|\mup - \mun|} ]
where (\sigmap) and (\sigman) are the standard deviations of the positive and negative controls, and (\mup) and (\mun) are their respective means [59].
Interpretation and Benchmarking: A Z'-factor ≥ 0.5 is typically considered excellent and indicates an assay with a robust separation band, ideal for HTS. Assays with 0 > Z'-factor < 0.5 are often deemed marginal but may be acceptable depending on the biological context and therapeutic need, particularly for more variable cell-based assays. A Z'-factor ≤ 0 suggests a low degree of separation, making the assay essentially unusable for screening purposes [56] [59]. It is crucial to distinguish Z'-factor from the Z-factor; the former uses only control data (no test samples) and is for assay validation, while the latter includes test samples and evaluates performance during or after a screen [56].
Signal-to-Background Ratio (S/B)
The Signal-to-Background Ratio (S/B) is a straightforward measure of an assay's dynamic range. It quantifies the fold difference between the mean signals of the positive and negative controls [57].
[ \text{S/B} = \frac{\mu{\text{signal}}}{\mu{\text{background}}} ]
A higher S/B ratio indicates a greater distinction between the positive and negative states of the assay, which facilitates the confident identification of active compounds (hits). While there is no universal threshold, a strong S/B is a fundamental requirement for achieving a high Z'-factor. It is often used in conjunction with other metrics, like signal-to-noise, to fully characterize the assay window [57].
Coefficient of Variation (CV)
The Coefficient of Variation (CV) standardizes the standard deviation of a set of measurements by expressing it as a percentage of the mean. This provides a relative measure of variability, or precision, that is independent of the unit of measurement, allowing for comparison across different assays or analyte concentrations [58].
[ \text{CV (%)} = \left( \frac{\text{Standard Deviation}}{\text{Mean}} \right) \times 100\% ]
In the context of assay validation, two types of CV are particularly important:
- Intra-assay CV: Measures precision within a single experiment or assay run. Target values are typically ≤ 5% for biochemical assays [58].
- Inter-assay CV: Measures precision across multiple independent experiments performed on different days. Target values are generally ≤ 10-15% [58].
The CV also has an operational interpretation; it can be used to calculate the probability that two replicate measurements of the same sample will differ by a factor of k or more, providing a direct link between precision and the likelihood of observing false disparities [60].
Table 1: Summary of Key Performance Metrics for Helicase Assay Validation
| Metric | Definition | Interpretation | Ideal Value | ||
|---|---|---|---|---|---|
| Z'-factor | ( 1 - \frac{3(\sigmap + \sigman)}{ | \mup - \mun | } ) | Measure of assay quality and separation band between controls. | ≥ 0.5 [56] [59] |
| Signal-to-Background (S/B) | ( \frac{\mu{signal}}{\mu{background}} ) | Measure of the dynamic range or assay window. | As high as possible; context-dependent [57] | ||
| Coefficient of Variation (CV) | ( \left( \frac{\text{Standard Deviation}}{\text{Mean}} \right) \times 100\% ) | Measure of precision (variability) (Intra-assay ≤ 5%, Inter-assay ≤ 10-15%) [58] |
Application in Helicase Activity Assays
Helicases, such as SARS-CoV-2 nsp13, are vital targets for antiviral and anticancer drug discovery. Screening for inhibitors typically relies on two primary biochemical activities: the direct unwinding of nucleic acid duplexes or the associated ATP hydrolysis [61] [62].
FRET-Based Unwinding Assay: As described for SARS-CoV-2 nsp13, this assay uses a nucleic acid substrate labeled with a fluorophore (e.g., Cy3) and a quencher. Upon helicase-mediated unwinding, the separation of strands causes a increase in fluorescence [61]. In this setup:
- Positive Control (High Signal): Contains the active helicase enzyme, ATP, and substrate, resulting in maximum unwinding and fluorescence.
- Negative Control (Low Signal): Lacks a key component (e.g., enzyme or ATP) or contains a denatured enzyme, yielding minimal fluorescence.
ATPase Activity Assay: This indirect assay detects ADP produced from helicase-catalyzed ATP hydrolysis using a homogeneous, "mix-and-read" method like Transcreener [62].
- Positive Control (High Signal): Contains active helicase with ATP and DNA/RNA cofactor, generating a high ADP signal.
- Negative Control (Low Signal): Lacks the helicase or uses an inactivated enzyme, showing baseline ADP levels.
For both assay types, these defined controls are used to calculate the Z'-factor, S/B, and CV, ensuring the assay is sufficiently robust before proceeding to screen compound libraries.
Diagram 1: Assay validation workflow.
Essential Reagents and Materials
The following table lists key reagents required for developing and validating a helicase activity assay, such as the FRET-based unwinding assay for SARS-CoV-2 nsp13 [61] [62].
Table 2: Research Reagent Solutions for Helicase Activity Screening
| Reagent/Material | Function in the Assay | Example/Note |
|---|---|---|
| Purified Helicase Enzyme | The catalytic target of the screening assay. | Must be nuclease-free and have a high percentage of active protein to avoid misinterpretation of results [63]. |
| Fluorescent Nucleic Acid Substrate | Serves as the reporter for unwinding activity. | A FRET-based DNA or RNA duplex with a fluorophore (e.g., Cy3) and a quencher (e.g., BHQ-2) [61]. |
| Nucleotide Cofactor | Provides energy for helicase activity. | ATP, used at a concentration near its Km value for the enzyme to ensure sensitivity [61]. |
| Reaction Buffer | Provides optimal pH, ionic strength, and cofactors. | Typically contains Mg²⁺, a pH buffer, salt (e.g., KCl), and DTT. Must be optimized for the specific helicase. |
| Positive Control | Generates the maximum assay signal. | A well with active helicase, substrate, and ATP. |
| Negative Control | Generates the minimum assay signal. | A well lacking helicase, ATP, or containing a heat-inactivated enzyme. |
| Microplates | The vessel for performing the HTS. | 384-well or 1536-well plates with low background fluorescence and good well-to-well uniformity. |
| Microplate Reader | Detects the fluorescence signal. | A reader capable of detecting the specific fluorophores used (e.g., FRET), ideally with HTS capabilities [56]. |
Detailed Experimental Protocol: FRET-Based Helicase Unwinding Assay
This protocol outlines the steps for performing a FRET-based helicase assay, similar to the one used for SARS-CoV-2 nsp13, including the subsequent calculation of performance metrics [61].
Reagent Preparation
- Helicase Assay Buffer: Prepare a buffer containing 25 mM HEPES (pH 7.5), 5 mM MgCl₂, 50 mM KCl, 1 mM DTT, and 0.01% Triton X-100. Filter sterilize and store at 4°C.
- Enzyme Solution: Dilute the purified, nuclease-free helicase in assay buffer to a final concentration of 3 nM (or as determined by titration). Keep on ice [61].
- Substrate Master Mix: Dilute the FRET-labeled DNA or RNA substrate in assay buffer. Include a 5-fold excess of an unlabeled DNA trap strand in the mix to prevent re-annealing of unwound strands [61].
- ATP Solution: Prepare a 10x ATP solution in assay buffer at a concentration of 1 mM (for a final concentration of 100 µM, near the Km) [61].
- Controls:
- Positive Control: Combine assay buffer, substrate mix, and ATP solution.
- Negative Control: Combine assay buffer, substrate mix, and an equivalent volume of water instead of ATP solution.
Assay Procedure for Validation
- Plate Layout: In a 384-well microplate, designate a minimum of 16 wells for the positive control and 16 wells for the negative control, distributed across the plate to assess spatial variability.
- Dispense Reagents:
- Add 10 µL of the Negative Control mix to the designated negative control wells.
- Add 10 µL of the Positive Control mix to the designated positive control wells.
- Initiating the Reaction:
- Using a multichannel pipette or dispenser, add 10 µL of the enzyme solution to all control wells.
- Centrifuge the plate briefly to mix and eliminate bubbles.
- Incubation and Reading:
- Incubate the plate at room temperature for 20 minutes, protected from light.
- Measure the fluorescence (excitation/emission for Cy3: ~550 nm/~570 nm) using a HTS-compatible microplate reader.
Data Analysis and Metric Calculation
- Calculate the mean ((\mup), (\mun)) and standard deviation ((\sigmap), (\sigman)) of the fluorescence readings for the positive and negative control wells.
- Compute the performance metrics using the following formulas:
- Z'-factor: ( Z' = 1 - \frac{3(\sigmap + \sigman)}{|\mup - \mun|} )
- S/B Ratio: ( S/B = \frac{\mup}{\mun} )
- CV (%): For positive controls: ( CVp = ( \sigmap / \mu_p ) \times 100\% ). Calculate similarly for negative controls.
Troubleshooting and Optimization Guidance
Assay validation may not meet targets on the first attempt. The diagram below outlines a logical framework for diagnosing and resolving common issues.
Diagram 2: Troubleshooting logic for assay optimization.
Rigorous assessment of the Z'-factor, Signal-to-Background ratio, and Coefficient of Variation is a non-negotiable step in the development of any semi-high-throughput helicase screening assay. These metrics collectively provide an objective, quantitative measure of an assay's robustness, dynamic range, and precision. By adhering to the protocols and interpretive frameworks outlined in this document, researchers can confidently validate their assay systems, ensuring that subsequent screening campaigns for helicase inhibitors are built upon a foundation of high-quality, reliable data, thereby accelerating the pace of discovery in antiviral and anticancer therapeutic development.
Within the context of semi-high-throughput screening for helicase activity research, the precise optimization of reaction conditions is a critical determinant of success. Helicases, molecular motors that unwind nucleic acids, are central to genomic maintenance and viral replication, making them prominent targets in drug discovery. This application note provides a detailed protocol and data-driven framework for optimizing three fundamental reaction components: magnesium, ATP, and substrate concentration. The goal is to establish robust and reproducible assay conditions suitable for the identification and characterization of helicase inhibitors, thereby accelerating therapeutic development.
Core Principles of Helicase Activity Assays
Helicases function as ATP-dependent enzymes that catalyze the separation of double-stranded nucleic acids into single strands. The energy derived from ATP hydrolysis is coupled to the mechanical work of unwinding. A common and efficient method for measuring this activity in a high-throughput format is to monitor the ATPase function indirectly by detecting the formation of ADP. The Transcreener ADP2 Assay is a homogenous, competitive immunoassay that utilizes a far-red fluorescent tracer and an antibody specific for ADP. As the helicase hydrolyzes ATP, the increasing concentration of ADP displaces the tracer from the antibody, resulting in a quantifiable signal change measurable by fluorescence polarization (FP), fluorescent intensity (FI), or time-resolved FRET (TR-FRET) [4] [64]. This universal detection method simplifies the protocol by eliminating the need for coupling enzymes and minimizes compound interference, making it ideal for high-throughput screening (HTS) [64].
Quantitative Optimization of Reaction Components
Systematic optimization is required to achieve maximal enzymatic activity and assay sensitivity. The following tables summarize key quantitative data and recommendations for magnesium, ATP, and nucleic acid substrates.
Table 1: Optimization of Magnesium and ATP Concentrations
| Helicase Enzyme | Optimal [Mg²⁺] | Optimal [ATP] | Key Observations & Considerations |
|---|---|---|---|
| SARS-CoV-2 nsP13 | 5 mM (at 3 mM ATP) [32] | 3 mM (for dsDNA); 9 mM (for dsRNA) [32] | Unhindered ATP hydrolysis is crucial for efficient translocation and unwinding. A slowly hydrolyzable ATP analog (ATPγS) inhibits unwinding [32]. |
| Human WRN | 10 mM (in 10X Buffer) [4] | Not explicitly specified | The provided 10X Enzyme Assay Buffer includes 10 mM MgCl₂. The final Mg²⁺ concentration in the reaction is 1 mM [4]. |
| Human DDX3, DDX5, DDX17 | 2 mM [64] | 100 µM (sub-Km used) [64] | Assayed in 50 mM Tris (pH 7.5), 2 mM MgCl₂, 0.01% Triton at 30 °C for 1 hour [64]. |
| Human RIG-I | 2.5 mM [64] | 100 µM (sub-Km used) [64] | Assayed in 50 mM Tris (pH 7.5), 50 mM NaCl, 2.5 mM MgCl₂, 0.01% Brij, 5 mM DTT at 30 °C for 1 hour [64]. |
| Human MDA5 | 2.5 mM (+ 1.25 mM MnCl₂) [64] | 100 µM (sub-Km used) [64] | Requires MnCl₂ as an additional cofactor in its optimized buffer [64]. |
Table 2: Optimization of Nucleic Acid Substrates
| Helicase Enzyme | Recommended Substrate | Substrate Concentration | Key Observations |
|---|---|---|---|
| SARS-CoV-2 nsP13 | dsDNA/RNA with a 5′-single-stranded tail [32] | Varies by experiment | Unwinds dsDNA more efficiently than dsRNA. A long 5′ ss-tail enhances dsDNA unwinding but can reduce dsRNA unwinding due to high affinity binding [32]. |
| Human WRN | 37-bp annealed 3′-Flap duplex DNA [4] | 40 µM stock [4] | The enzyme has 3'→5' helicase activity and exonuclease activity towards double-stranded DNA with a 5'-overhang [4]. |
| Human DDX3, DDX5, DDX17 | Yeast RNA (common substrate) [64] | 1 mg/mL (saturating) [64] | Yeast RNA was selected as a suitable common substrate for all three DDX enzymes to standardize assay conditions [64]. |
| Human RIG-I | 5′ppp dsRNA [64] | 0.2 ng/μL [64] | This specific viral RNA substrate is recognized by RIG-I for innate immune activation [64]. |
| Human MDA5 | High Molecular Weight (HMW) poly(I:C) [64] | 2 ng/μL [64] | HMW poly(I:C), a double-stranded RNA analog, is the preferred substrate for activating MDA5 [64]. |
Detailed Experimental Protocols
Protocol: Helicase ATPase Activity Assay using Transcreener ADP2
This protocol is configured for a 10 µL enzyme reaction in a 384-well plate format [4] [64].
Materials Required:
- Enzyme: Purified helicase (e.g., WRN, DDX3, RIG-I).
- Substrate: Appropriate nucleic acid (see Table 2).
- Cofactors: MgCl₂, ATP, DTT (if required).
- Assay Buffer: Tris-HCl or HEPES buffer, pH 7.5, with detergent.
- Detection Kit: Transcreener ADP2 Assay Kit (FP, FI, or TR-FRET format).
- Equipment: Multimode microplate reader, liquid handling devices, 384-well low-volume assay plates.
Procedure:
- Preparation of Reaction Master Mix:
- Prepare a master mix on ice containing the following components to the indicated final concentrations:
- 1X Enzyme Assay Buffer (e.g., 50 mM Tris-HCl pH 7.5, 2 mM MgCl₂, 0.01% Triton) [64].
- ATP (typically 100 µM for sub-Km conditions or higher if needed for full unwinding) [32] [64].
- Nucleic Acid Substrate at the optimized concentration (see Table 2).
- DTT (if required for enzyme stability, e.g., 5 mM for RIG-I) [64].
- Dispense the master mix into the designated wells of a 384-well assay plate.
- Prepare a master mix on ice containing the following components to the indicated final concentrations:
Initiating the Enzymatic Reaction:
- Start the reaction by adding the purified helicase enzyme. The enzyme concentration must be determined empirically to ensure activity is within the linear range (e.g., 0.9 nM for RIG-I, 50 nM for DDX3) [64].
- Seal the plate to prevent evaporation and incubate at the recommended temperature (typically 30°C) for the predetermined time (e.g., 1-2 hours) [64].
Stopping the Reaction and Detecting ADP:
- After the incubation, stop the reaction by adding an equal volume (e.g., 10 µL) of Stop & Detect Buffer B (2X). This buffer contains EDTA, which chelates Mg²⁺ and halts all enzymatic activity [4].
- Simultaneously, this mixture introduces the ADP2 Antibody and the Fluorescent Tracer specific to your chosen detection format (FP, FI, or TR-FRET).
Signal Measurement and Data Analysis:
- Incubate the plate at room temperature for the time specified by the Transcreener protocol to allow for signal development.
- Read the plate using the appropriate settings on your multimode plate reader.
- Generate an ADP standard curve to convert the raw fluorescence values into moles of ADP produced. Calculate enzymatic velocities and determine the effects of inhibitors using IC₅₀ curves [64].
Workflow Diagram: Helicase ATPase Activity Screening
The following diagram illustrates the key steps and decision points in the semi-high-throughput screening workflow.
Mechanism Diagram: ADP Detection with Transcreener
This diagram outlines the fundamental competitive immunoassay principle of the Transcreener ADP2 Assay.
The Scientist's Toolkit: Research Reagent Solutions
A successful helicase screening campaign relies on high-quality, well-characterized reagents. The following table details essential components and their functions.
Table 3: Essential Research Reagents for Helicase Assay Development
| Reagent / Solution | Function & Role in Assay | Example & Notes |
|---|---|---|
| Transcreener ADP2 Assay Kits | Universal, antibody-based detection of ADP for measuring ATPase activity. Available in FP, FI, and TR-FRET formats [4] [64]. | BellBrook Labs (Cat# 3010-1K, 3013-1K, 3011-1K). Enables homogenous, "mix-and-read" detection suitable for HTS [4]. |
| Enzolution WRN Helicase ATPase Assay System | A comprehensive kit providing pre-optimized reagents for a specific helicase, including enzyme and substrate [4]. | Includes WRN Helicase (0.1 mg/mL), 37-bp 3'-Flap DNA substrate, and 10X Enzyme Assay Buffer [4]. |
| Purified Recombinant Helicases | The enzyme target for screening and mechanistic studies. Requires high purity and activity. | Examples: SARS-CoV-2 nsP13 [32], human DDX1, MDA5, LGP2 [65]. Often expressed with affinity tags (e.g., 6xHis) for purification. |
| Defined Nucleic Acid Substrates | Act as the physiological stimulus for the helicase's ATPase and unwinding activities. | Examples: 5'ppp dsRNA for RIG-I, HMW poly(I:C) for MDA5, yeast RNA for DDX enzymes, tailed duplex DNA for nsP13 [32] [64]. |
| Stop & Detect Buffer B | A critical component that quenches the enzymatic reaction and initiates detection. | Contains EDTA to chelate Mg²⁺, stopping the reaction, and is formulated for use with the detection antibody and tracer [4]. |
In the context of semi-high-throughput helicase activity screening, differentiating true enzymatic inhibition from systematic assay artifacts is a critical challenge. Compound interference arises when test molecules affect the assay detection system rather than the target biology, leading to false positives that can misdirect research and drug development efforts. These artifacts are particularly problematic in high-throughput screening (HTS), where genuine active compounds are rare (typically 0.01–0.1% of a library) and easily obscured by false signals [66]. This application note details the origins of these artifacts and provides validated protocols to mitigate them, specifically focusing on helicase activity assays.
Origins and Mechanisms of Assay Interference
Understanding the biological and chemical origins of interference is essential for developing effective countermeasures. The following table summarizes the primary mechanisms of compound interference relevant to helicase assays.
Table 1: Common Mechanisms of Compound Interference in Biochemical Assays
| Interference Type | Mechanism of Action | Impact on Assay Readout |
|---|---|---|
| Compound Fluorescence [66] [67] | Compounds with conjugated bonds absorb and emit light at wavelengths overlapping the assay's detection window. | False positive signals in fluorescence-based detection (e.g., increased signal). |
| Signal Quenching [68] [67] | Compounds absorb excitation or emission light, attenuating the fluorescent signal. | False negative signals or underestimated activity (e.g., decreased signal). |
| Compound Aggregation [66] | Molecules self-associate to form colloidal aggregates (50–400 nm) that non-specifically sequester enzymes. | Promiscuous inhibition leading to false positives; often enzyme-dependent. |
| Redox Cycling [66] | Compounds (e.g., quinones) generate reactive oxygen species in the presence of reducing agents like DTT. | Artificial modulation of signal, leading to false positives or negatives. |
| Enzyme-Specific Inhibition [66] | Direct inhibition of reporter enzymes (e.g., firefly luciferase) used in coupled systems. | False positives that are reproducible and concentration-dependent. |
Experimental Protocols for Mitigation and Validation
Protocol: Orthogonal Assay for Hit Confirmation
This protocol uses a secondary assay with a different detection mechanism to validate primary screen hits, effectively ruling out technology-specific artifacts [68].
- Primary Screening: Conduct the initial helicase screen using your standard method (e.g., the molecular beacon assay detailed in Protocol 3.2).
- Hit Identification: Select compounds that show activity above the defined threshold (e.g., >50% inhibition).
- Orthogonal Assay:
- Principle: Confirm hits using an assay that detects a different product or uses a different physical principle. For a helicase assay that uses fluorescence polarization, an orthogonal assay could be a luminescence-based or radiometric format [68].
- Example - Radiometric Gel-Based Assay: As described in [8], this traditional method involves: a. Substrate Preparation: Anneal a radio-labeled (e.g., γ-32P) oligonucleotide to a complementary strand to form a duplex. b. Reaction: Incubate the helicase with the substrate and ATP in an appropriate buffer. c. Termination & Separation: Stop the reaction with a stop solution (e.g., containing SDS and excess unlabeled trap oligonucleotide to prevent re-annealing). Resolve the reaction products on a non-denaturing polyacrylamide gel. d. Visualization & Quantification: Visualize and quantify the separated single-stranded (unwound) and double-stranded (substrate) DNA using a phosphorimager.
- Data Analysis: Compare the dose-response curves from the primary and orthogonal assays. True inhibitors will show congruent activity in both formats, while interferers will not.
Protocol: Molecular Beacon-Based Helicase Assay
This protocol describes a continuous, homogeneous fluorescence assay that minimizes common artifacts like product re-annealing and is amenable to HTS [8].
Principle: A molecular beacon—a single-stranded DNA oligonucleotide with a fluorophore and a quencher on each end—is annealed to a longer complementary strand. Helicase unwinding releases the beacon, which then forms an intramolecular hairpin, bringing the quencher and fluorophore together and reducing fluorescence. The irreversibility of hairpin formation eliminates the need for single-stranded DNA traps.
Materials:
- Recombinant helicase (e.g., HCV NS3 helicase domain)
- DNA or RNA oligonucleotides (see Table 2 for sequences)
- Fluorescently-labeled molecular beacon (e.g., Cy3-IBRQ or Cy5-BHQ)
- Assay Buffer: 25 mM MOPS pH 6.5, 2 mM MgCl₂
- ATP solution (0.5 M, pH 7.0)
- White half-volume 96- or 384-well microplates
- Fluorescence plate reader with temperature control
Procedure:
- Substrate Preparation: Anneal the molecular beacon to its complementary strand at a 1:1 molar ratio (e.g., 20 µM each) in 10 mM Tris-HCl, pH 8.5. Heat to 95°C for 5 minutes and allow to cool slowly to room temperature (~1 hour).
- Assay Setup: In a white half-volume microplate, add:
- 25 nM Helicase enzyme
- 5 nM Annealed substrate
- Test compound or control (DMSO concentration should be normalized, typically <1%)
- Assay Buffer to a final volume of 100 µL.
- Signal Acquisition: Initiate the reaction by adding ATP to a final concentration of 0.5 mM. Immediately place the plate in the reader and monitor fluorescence every 30-60 seconds for 30-60 minutes at 22°C.
- Cy3-labeled beacon: Ex/Em = 552/570 nm
- Cy5-labeled beacon: Ex/Em = 643/667 nm
- Data Analysis: Fit the fluorescence decay over time to a first-order exponential decay model to determine the observed rate constant (kobs) for each reaction.
Protocol: Counter-Screen for Identifying Promiscuous Inhibitors
This protocol identifies compounds that act through non-specific mechanisms like aggregation [68] [66].
- Primary Screen: Identify active compounds from the primary helicase screen.
- Aggregation Counter-Screen:
- Principle: Non-ionic detergents can disrupt compound aggregates.
- Method: Re-test all primary hits in the standard helicase assay in the presence and absence of a non-ionic detergent (e.g., 0.01% Triton X-100 or Tween-20).
- Interpretation: A significant reduction (e.g., >50%) in inhibitory activity in the presence of detergent suggests the compound was acting via aggregation.
- Enzyme-Free Counter-Screen:
- Principle: Measures the intrinsic effect of the compound on the assay signal.
- Method: Run the assay in the absence of the helicase enzyme. Include the substrate, detection reagents, and test compound.
- Interpretation: Compounds that significantly alter the baseline signal in this counter-screen are likely fluorescent or quench the signal.
- Data Triage: Compounds that fail these counter-screens should be deprioritized or studied further with great caution.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Key Reagents for Robust Helicase Assays
| Reagent / Material | Function / Rationale | Example & Specification |
|---|---|---|
| Non-Ionic Detergent [66] | Disrupts compound aggregates, reducing promiscuous inhibition. | Triton X-100 or Tween-20, used at 0.01% v/v in assay buffer. |
| Molecular Beacon Substrate [8] | Provides an irreversible, homogeneous, and continuous readout of helicase activity, minimizing re-annealing artifacts. | Cy3-/5'-IBRQ/BHQ-labeled DNA; sequence: 5'-/IBRQ/AGTGCGCTGTATCGTCAAGGCACT/Cy3/-3'. |
| Orthogonal Detection Reagents [68] | Enables hit confirmation via a different physical principle (e.g., radiometric, luminescence). | Radio-labeled (γ-32P) nucleotides for gel-shift assays. |
| Reducing Agent Alternatives [66] | Replaces DTT/TCEP to avoid redox cycling artifacts with susceptible compounds. | Consider lower concentrations or alternative stabilizers if enzyme activity permits. |
| High-Quality Enzyme Preps [68] | Minimizes lot-to-lot variability and ensures consistent, specific activity. | Recombinant helicase (e.g., HCV NS3); aliquot to avoid freeze-thaw cycles; use validated suppliers. |
Data Analysis and Quality Control
Robust data analysis is critical for distinguishing true signals from noise. The Z' factor is a key metric for assessing assay quality and robustness in HTS [68].
- Calculation: Z' = 1 - [ (3σ₊ + 3σ₋) / |μ₊ - μ₋| ] where σ₊ and σ₋ are the standard deviations of positive and negative controls, and μ₊ and μ₋ are their means.
- Interpretation: A Z' factor ≥ 0.5 is considered an excellent assay suitable for HTS. Assays with Z' ≥ 0.7 exhibit a large separation between controls and are highly robust [68].
Table 3: Quantitative Impact of Mitigation Strategies on Assay Performance
| Strategy | Parameter Measured | Impact on Data Quality |
|---|---|---|
| Addition of Detergent [66] | Reduction in aggregate-based false positives. | Can reduce promiscuous hit rates by >50%. |
| "Red-Shifted" Detection [67] | Percentage of fluorescent compounds in a library. | Fluorescent compounds drop from ~5% (UV/blue) to 0.01-0.1% (far-red). |
| Orthogonal Confirmation [68] | Hit confirmation rate. | Can eliminate >80% of fluorescence-based false positives [67]. |
| Optimized Reagent Quality [68] | Inter-assay coefficient of variation (CV). | Reduces CV, improving reproducibility and Z' factor. |
Strategies for Enzyme Stability and Reagent Storage to Ensure Reproducibility
Enzyme stability is a cornerstone of reproducibility in biochemical research, particularly in semi-high-throughput screening (HTS) for helicase activity. Enzymes are inherently unstable molecules; their catalytic activity depends on the preservation of a precise three-dimensional structure, which is susceptible to degradation from factors such as temperature, pH, and storage conditions [69]. In the context of drug discovery, where helicase enzymes are emerging therapeutic targets for cancer, antiviral therapies, and autoimmune diseases, inconsistent enzymatic performance can lead to inaccurate data, failed experiments, and costly delays in lead optimization [70]. This application note details evidence-based strategies for enzyme and reagent storage, alongside standardized protocols, to ensure the reliability and reproducibility of semi-high-throughput helicase activity assays.
Understanding Enzyme Instability
Enzyme instability can be categorized into two major types: shelf (storage) stability, which refers to the retention of activity over time when stored, and stability in operations, which relates to the retention of activity during use [71]. The primary challenge is that enzymes possess low inherent stability due to a complex balance of intramolecular forces and interactions with the surrounding solvent [71]. Key factors contributing to instability include:
- Temperature: Elevated temperatures accelerate denaturation and bacterial growth, leading to premature degradation. Most enzymes gradually lose catalytic activity when stored at temperatures higher than recommended [69].
- Hydration State: Enzymes in aqueous solutions are unstable due to various intramolecular and intermolecular chemical reactions, including hydrolysis, aggregation, deamidation, and oxidation [72].
- Mechanical and Chemical Stress: Processes like freezing and drying, exposure to organic solvents, or high-pressure treatments can induce conformational changes or aggregation, resulting in activity loss [72] [71].
For helicase enzymes—molecular motors that unwind nucleic acids using ATP hydrolysis—maintaining stability is critical for accurate measurement of both ATPase and unwinding activities in HTS workflows [70].
Best Practices for Enzyme Storage and Stabilization
Temperature and Storage Conditions
The fundamental rule for enzyme storage is maintaining a consistent, low temperature. However, a one-size-fits-all approach does not apply.
Table 1: Recommended Storage Temperatures for Different Enzyme Types
| Enzyme Type / Context | Recommended Storage Temperature | Key Considerations |
|---|---|---|
| General Enzyme Storage [69] | -20°C | Often combined with glycerol to prevent protein denaturation. |
| Oxidoreductases [69] | Varies; can be complex | Require optimized cold storage; specific needs depend on the enzyme. |
| Transferases [69] | -70°C | Clinical studies show this temperature guarantees the best stability. |
| Hydrolases & Isomerases [69] | -20°C | Dilution in a buffered solution at this temperature can retain activity for years. |
| Lyases (Long-Term) [69] | -80°C | Ultra-low freezer storage is optimal, often with stabilizing reagents. |
| Enzymes in Sequencing Kits [73] | 2–8°C (post-thaw) | Thawed reagent cartridges are stable for 6 hours on ice or up to 1 week if thawed overnight at 2–8°C. Refreezing is not recommended. |
For long-term storage, ultra-low temperature freezers (-70°C to -80°C) are often required. High-performance laboratory-grade refrigerators and freezers are designed to minimize temperature variances and ward off humidity, which is crucial for preserving sensitive enzymes [69].
Formulation and Additives
The formulation of the enzyme product is equally as important as the storage temperature.
- Liquid vs. Solid Formulations: A study on methyl trypsin demonstrated that reformulating a liquid enzyme into a granulate significantly improved stability. The original liquid-liquid formulation was stable for only 1 day at room temperature after reconstitution, whereas the solid-liquid granulate was stable for up to 3 days [72]. This highlights that maintaining enzymes in a solid form until reconstitution can dramatically enhance shelf-life.
- Use of Stabilizing Agents: The addition of glycerol (often at 50% v/v) is a widely accepted practice to prevent protein denaturation at -20°C [69]. Other successful strategies include storing peroxidase-labeled immunoglobulins as ammonium sulfate precipitates at 4°C, which allowed for the retention of over 90% of both enzymatic and immunological activity after two years [69].
- Optimized Buffers: Enzymes should be diluted or stored in appropriate buffered solutions to maintain pH and ionic strength, which are critical for preserving the enzyme's native conformation [69].
Reagent Storage and Handling for Helicase Assays
Semi-high-throughput helicase screening relies on a suite of specialized reagents, whose stability directly impacts assay reproducibility.
Table 2: Research Reagent Solutions for Helicase Screening
| Reagent / Material | Function in Helicase Assay | Storage & Handling Considerations |
|---|---|---|
| Purified Helicase Enzyme (e.g., DDX3, WRN, BLM) [70] | The catalytic target; unwinds nucleic acid duplexes. | Store at -70°C to -80°C. Avoid repeated freeze-thaw cycles. Use stabilizing buffers with glycerol. |
| Transcreener ADP² Assay [70] [64] | Homogeneous, "mix-and-read" assay to detect ADP produced from ATP hydrolysis. | Follow manufacturer's guidelines. The antibody and tracer are typically stored at -20°C. |
| Nucleic Acid Substrates (e.g., yeast RNA, Poly(I:C)) [64] | The substrate unwound by the helicase; essential for activity. | Store frozen aliquots to avoid nuclease degradation. Avoid repeated freezing and thawing. |
| ATP [74] | The energy source for the helicase's unwinding activity. | Store in stable, frozen aliquots at -20°C. Avoid acidic conditions that promote hydrolysis. |
Critical Handling Procedures
- Thawing Protocols: For sensitive enzyme-containing reagents, such as those in Illumina MiSeq kits, two thawing methods are acceptable: a room temperature water bath for 60-90 minutes (use within 6 hours) or an overnight thaw in a refrigerator (2-8°C) (stable for up to 1 week) [73]. The latter method offers greater flexibility for assay planning.
- Avoiding Refreezing: Unless specified, refreezing thawed reagents is not recommended as it can negatively impact enzyme activity and overall performance [73].
- Aliquoting: To minimize repeated freeze-thaw cycles, which are detrimental to enzyme activity, reagents and enzymes should be aliquoted into single-use volumes upon arrival.
Experimental Protocols for Stability Assessment and Screening
Protocol: Helicase ATPase Activity Assay (ADP Detection)
This universal protocol is ideal for primary high-throughput screening of helicase inhibitors, as it measures the fundamental ATP hydrolysis event [70] [64].
Principle: The helicase enzyme hydrolyzes ATP to ADP in the presence of its RNA/DNA substrate. The Transcreener ADP² assay uses a highly specific antibody to detect the produced ADP. The displacement of a fluorescent tracer from the antibody results in a measurable signal change (Fluorescence Polarization, FP; Fluorescence Intensity, FI; or Time-Resolved FRET, TR-FRET) [64].
Workflow of Helicase ATPase Activity Assay
Materials:
- Purified helicase enzyme (e.g., DDX3, RIG-I)
- Transcreener ADP² Assay Kit (BellBrook Labs)
- RNA substrate (e.g., yeast RNA for DDX enzymes, 5'ppp dsRNA for RIG-I)
- ATP
- Assay buffer (e.g., 50 mM Tris pH 7.5, 2-2.5 mM MgCl₂, 0.01% detergent)
- 384-well or 1536-well microplates
Procedure:
- Prepare Reaction Mix: In a low-volume microplate, combine the helicase enzyme with ATP (e.g., 100 µM, a sub-Km concentration) in the appropriate assay buffer [64].
- Initiate Reaction: Start the enzymatic reaction by adding the required RNA substrate (e.g., 1 mg/mL yeast RNA for DDX3) [64].
- Incubate: Incubate the reaction at 30°C for 1-2 hours to allow for initial velocity conditions [64].
- Detect ADP: Stop the reaction by adding the Transcreener ADP² detection mixture (containing the antibody and tracer).
- Signal Development: Incubate the plate at room temperature for 30-60 minutes to allow for signal development.
- Read Plate: Measure the signal using a plate reader configured for FP, FI, or TR-FRET.
- Data Analysis: Convert raw fluorescence values to ADP concentrations using a standard curve. Calculate reaction rates and Z' factors to validate assay robustness for HTS (Z' ≥ 0.7 is considered excellent) [64].
Protocol: Orthogonal Validation with a Fluorescent Unwinding Assay
While ATPase assays are excellent for primary screening, they are indirect. Confirmation of true helicase inhibition requires an orthogonal assay that directly measures strand separation [70] [74].
Principle: Two fluorescently modified oligonucleotides are annealed to an unmodified loading strand. One strand is labeled with a fluorophore (e.g., Cy3), the other with a quencher. When the duplex is intact, fluorescence is quenched. Helicase-catalyzed unwinding separates the strands, leading to an increase in fluorescence that can be monitored in real-time [74].
Materials:
- Purified helicase enzyme
- Fluorescently labeled duplex substrate
- ATP
- Reaction buffer (with Mg²⁺)
- Real-time PCR machine or fluorescence plate reader
Procedure:
- Prepare Substrate: Anneal the fluorophore-labeled and quencher-labeled oligonucleotides to the longer, unmodified loading strand to form the duplex substrate.
- Setup Reaction: In a 96- or 384-well plate, mix the helicase with the duplex substrate and ATP in reaction buffer.
- Real-Time Measurement: Immediately place the plate in a real-time thermocycler or fluorometer pre-heated to the reaction temperature (e.g., 30°C or 37°C). Monitor the fluorescence increase over time (e.g., 1-2 hours).
- Data Analysis: Calculate unwinding rates from the fluorescence trajectories. This setup allows for precise temperature control and multiplexing with different fluorophores [75].
Stability Testing and Quality Control
To ensure long-term reproducibility, a systematic approach to stability testing is required.
- Storage Stability Studies: Conduct long-term stability studies in accordance with ICH guidelines, storing enzyme formulations at various temperatures (e.g., 5°C, 25°C/40% RH, 30°C/60% RH) and assaying for proteolytic activity at set time points (e.g., 0, 3, 6, 9, 12, 18, and 24 months) [72].
- Defining Half-Life: Enzyme stability is often expressed as its half-life, the time taken for enzyme activity to fall to half of its original value [71].
- Activity Assays: Regularly monitor enzyme activity using standardized assays, such as a spectrophotometric proteolytic activity assay for proteases [72]. For helicases, this involves running control unwinding or ATPase reactions with reference inhibitors (e.g., Suramin) to ensure consistent performance and calculate IC₅₀ values for validation [64].
The reproducibility of semi-high-throughput helicase screening assays is inextricably linked to rigorous strategies for enzyme stability and reagent storage. By implementing the core practices outlined in this document—utilizing appropriate, consistent storage temperatures, employing stabilizing formulations, adhering to strict reagent handling protocols, and validating assays with robust, orthogonal methods—researchers can significantly reduce experimental variability. This disciplined approach provides a solid foundation for generating reliable, high-quality data, thereby accelerating the discovery of novel helicase inhibitors in oncology, virology, and beyond.
From Hits to Leads: Orthogonal Assays and Mechanistic Validation
In the development of semi-high-throughput screening assays for helicase activity, reliance on a single analytical method introduces significant risk. False positives, assay artifacts, or overlooked off-target effects can compromise data integrity and lead to costly late-stage failures in drug discovery pipelines. An orthogonal approach—employing multiple, independent methods to measure the same biological endpoint—is therefore critical for robust results. This strategy validates findings through differing physical or chemical principles, ensuring that observed activities are genuine and not artifacts of a particular assay format [76] [77]. For helicase research, integrating a gel-based assay, which provides direct visualization of nucleic acid substrate displacement, with a cell-based confirmation, which confirms activity in a physiologically relevant environment, creates a powerful synergy. This application note details a structured orthogonal strategy, combining these methodologies to deliver high-confidence validation of helicase function and inhibition, specifically framed within a semi-high-throughput screening context.
Orthogonal Assay Design and Workflow
The core of the orthogonal strategy is a two-tiered system designed for efficiency and confidence. The primary, high-throughput tier utilizes a homogeneous, fluorescent-based assay to rapidly screen compound libraries for potential helicase inhibitors or activators. This is immediately followed by a secondary, confirmatory tier employing the orthogonal gel-based assay to visually confirm hits from the primary screen. Finally, promising candidates advance to a cell-based assay to evaluate their activity and specificity within a complex cellular environment [75] [4].
The following workflow diagram outlines the sequential integration of these independent methods:
Experimental Protocols
Semi-High-Throughput Fluorescent Primary Screen
This protocol is adapted for a 384-well plate format and is designed to detect ADP formation as a measure of helicase ATPase activity, which correlates with unwinding function [4].
Materials & Reagents
Table 1: Key Reagents for Fluorescent Helicase ATPase Assay
| Reagent | Function/Description | Source/Example |
|---|---|---|
| Purified Helicase (e.g., WRN 500-946) | Catalytic enzyme component; ATP-dependent unwinding activity. | BellBrook Labs [4] |
| DNA Substrate (e.g., 37-bp 3'-Flap duplex) | Helicase nucleic acid target; its unwinding consumes ATP. | BellBrook Labs [4] |
| Transcreener ADP2 Assay Kit | Homogeneous, mix-and-read detection of ADP formation via competitive fluorescence immunoassay. | BellBrook Labs [4] |
| ATP | Energy substrate for the helicase reaction. | Kit provided [4] |
| 10X Enzyme Assay Buffer | Provides optimal pH and cofactors (e.g., Mg²⁺). | Kit provided [4] |
| 384-Well Low Volume Assay Plates | Optimized for fluorescent readings and minimal reagent use. | Corning #4514 [4] |
Step-by-Step Procedure
Reaction Setup: In a 384-well plate, combine the following in a final reaction volume of 10 µL per well:
- 2 µL of test compound or control (DMSO for negative control, known inhibitor for positive control).
- 2 µL of DNA substrate (at a final reaction concentration of 200-500 nM).
- 4 µL of helicase enzyme (diluted in 1X Enzyme Assay Buffer to a predetermined optimal concentration).
- 2 µL of ATP (at a final reaction concentration of 10-50 µM).
Incubation: Seal the plate and incubate at 30°C for 60 minutes to allow the enzymatic reaction to proceed.
Detection: Terminate the reaction by adding 10 µL of Stop & Detect Buffer containing the fluorescent tracer and antibody from the Transcreener ADP2 kit. The EDTA in this buffer chelates Mg²⁺, stopping the enzyme reaction.
Reading: Incubate the plate for a further 10-60 minutes at room temperature and then measure the fluorescence signal (FP, FI, or TR-FRET) using a compatible multimode plate reader.
Orthogonal Gel-Based Helicase Assay
This method provides direct, visual proof of nucleic acid strand displacement by separating the reaction products via native polyacrylamide gel electrophoresis (PAGE) [75].
Materials & Reagents
- Radioactive or Fluorescently-Labeled DNA Substrate: A forked duplex or partial duplex DNA with a strand labeled for detection.
- Helicase Assay Buffer: Typically containing Tris-HCl (pH 7.5-8.0), MgCl₂, NaCl, DTT, and ATP.
- Stop Solution: 50-100 mM EDTA, 40% glycerol, 0.6% SDS, and 0.1% bromophenol blue.
- Equipment: Gel electrophoresis apparatus, phosphorimager or fluorescence gel scanner.
Step-by-Step Procedure
Reaction Assembly: On ice, set up 20 µL reactions containing:
- 1X Helicase Assay Buffer.
- Labeled DNA substrate (10-50 nM).
- Purified helicase enzyme.
- Test compound or control.
- Initiate the reaction by adding ATP to a final concentration of 2-5 mM.
Incubation: Transfer reactions to a 37°C heat block and incubate for 30-60 minutes.
Reaction Termination: Add 5 µL of stop solution to each reaction to chelate Mg²⁺ and halt enzyme activity.
Product Separation: Load the entire reaction onto a pre-run, non-denaturing polyacrylamide gel (e.g., 10-12%) in 0.5X TBE buffer. Run the gel at 80-100 V for 60-90 minutes with cooling to maintain a constant temperature.
Visualization and Quantification: Expose the gel using a phosphorimager or fluorescence scanner. Quantify the bands corresponding to the substrate (double-stranded) and product (single-stranded) using image analysis software (e.g., ImageJ). Calculate the percent unwinding using the formula:
% Unwinding = (Product Signal / (Product Signal + Substrate Signal)) * 100.
Cell-Based Confirmation Assay
This protocol leverages a synthetic biology approach to confirm helicase-targeting activity in a live-cell context [78].
Materials & Reagents
- Engineered Cell Line: T-cells or a mammalian cell line (e.g., HEK293) stably expressing a synthetic receptor (e.g., NatE MESA) designed to sense a specific cue and, upon activation, express a reporter gene (e.g., GFP, luciferase).
- Cell Culture Media: Appropriate medium (e.g., RPMI-1640 for T-cells, DMEM for HEK293) supplemented with serum and antibiotics.
- Reporter Lysis Buffer: For luciferase-based assays.
Step-by-Step Procedure
Cell Seeding: Seed engineered cells into a 96-well or 384-well tissue culture plate at a density of 10,000-50,000 cells per well.
Compound Treatment: Add the confirmed hits from the gel-based assay to the cells. Include controls (vehicle and a positive control if available).
Incubation: Incubate the cells for 24-48 hours in a 37°C, 5% CO₂ incubator to allow for synthetic receptor signaling and reporter gene expression.
Reporter Detection:
- For Fluorescent Reporters (e.g., GFP): Directly measure fluorescence intensity with a plate reader.
- For Luciferase Reporters: Lyse the cells following the incubation period and add the luciferase substrate. Measure the luminescent signal.
Data Analysis: Normalize the signal from treated wells to vehicle control wells. A significant increase in reporter signal indicates that the compound is active in the cellular environment and can engage the intended pathway.
Data Interpretation and Validation
The power of the orthogonal strategy is realized when data from all three assays are integrated. The following table summarizes the key attributes and performance metrics for each method:
Table 2: Orthogonal Assay Comparison and Performance Metrics
| Assay Parameter | Fluorescent Primary Screen | Gel-Based Assay | Cell-Based Confirmation |
|---|---|---|---|
| Throughput | High (384-well) | Medium (96-well format) | Low (96-well) |
| Readout | ADP formation (Indirect) | Nucleic acid strand displacement (Direct) | Reporter gene expression (Functional) |
| Key Performance Metric | Z'-factor > 0.5, Signal-to-Noise > 10 | % Unwinding, IC₅₀ | Fold induction over control, EC₅₀/IC₅₀ |
| Quantitative Data Output | IC₅₀ from dose-response curves | Direct quantification of substrate and product bands | Dose-response curves for cellular activity |
| Role in Workflow | Triage large libraries | Confirm mechanism and specificity of primary hits | Validate physiological activity and specificity |
Assay Validation and Quality Control
For any quantitative assay, proper analytical validation is essential to ensure reliability. Key parameters to establish include [79]:
- Precision: Determine intra-assay and inter-assay variability using a minimum of 2 concentrations of quality control material tested in duplicate over 20 days. The total observed variance should not exceed 33% of the total allowable error.
- Accuracy/Bias: Compare the assay results to a reference method using at least 40 patient or sample measurements. The mean bias should be less than half of the total allowable error.
- Linearity: Verify the assay's analytical measuring range using at least 5 sample pools tested in duplicate. The results should demonstrate a linear relationship between expected and observed values.
The presented orthogonal strategy, which seamlessly integrates a semi-high-throughput fluorescent screen with gel-based confirmation and cell-based functional analysis, provides a robust framework for helicase research and drug discovery. This multi-faceted approach mitigates the risk of false positives inherent in single-assay systems and delivers high-confidence data on compound efficacy and mechanism of action. By adopting this comprehensive workflow, researchers can accelerate the identification and validation of novel helicase modulators with greater certainty, ultimately enhancing the efficiency of the drug development pipeline.
Counterscreening to Rule Out Non-Specific Inhibition and DNA Damage
In semi-high-throughput helicase activity screening assays, the initial identification of "hit" compounds is only the first step. A significant challenge in helicase drug discovery is that primary screening hits often include compounds that act through non-specific mechanisms rather than directly and specifically inhibiting the target helicase. These non-specific mechanisms include interference with assay detection systems, general DNA or RNA damage, and disruption of essential cofactors. Such artifacts can lead to false positives that waste resources and misdirect research efforts. Therefore, implementing robust counterscreening strategies is essential to distinguish true helicase inhibitors from non-specific compounds [80].
This application note details standardized protocols for counterscreening assays designed to identify and eliminate non-specific inhibitors and DNA-damaging compounds from helicase drug discovery campaigns. We focus on practical, semi-high-throughput methods that can be implemented alongside primary screening to efficiently triage hits and advance only the most promising candidates for further development.
Key Counterscreening Strategies & Case Studies
Ribozyme Interference Assay for Specificity Validation
Background & Principle: The Ribozyme Insertion Deletion Editing (RIDE) assay was developed to identify inhibitors of the kinetoplastid RNA editing pathway but provides an excellent model for specificity testing in nucleic acid enzyme screens. This approach validates whether inhibition observed in the primary assay is specific to the target enzyme rather than resulting from interference with common assay components like reporter systems [81].
Protocol Insights:
- Assay Configuration: The core principle involves testing candidate inhibitors in a simplified system that maintains the detection chemistry but removes the primary enzymatic target. For helicase assays, this translates to using the same fluorescent or luminescent reporter system but without the helicase protein.
- Procedure: Prepare reaction mixtures identical to your primary helicase assay but omit the helicase enzyme. Add candidate inhibitors at the same concentrations used in primary screening. Incubate under the same conditions (time, temperature) as the primary assay.
- Data Interpretation: Compounds showing significant "inhibition" in this target-free system are flagged as non-specific interferers. In the RIDE assay example, 5 of 10 initial hits were eliminated through this process, including anthracycline compounds like doxorubicin and daunorubicin that interfered with ribozyme activity or fluorescence detection [81].
Application to Helicase Research: Adapt this approach by creating a helicase-free version of your primary screening assay. For fluorescence-based unwinding assays, this would include all detection components (fluorescently-labeled substrates, quenchers, dyes) without the helicase. Signal reduction indicates direct interaction with the nucleic acid substrate or interference with the detection chemistry.
Orthogonal Assay with Alternative Detection Mechanism
Background & Principle: The SARS-CoV-2 nsP13 helicase screening campaign implemented an orthogonal counterscreen using a different detection methodology to confirm true inhibitors. This approach identifies compounds that inhibit regardless of detection technology, increasing confidence in hit validity [30].
Case Study Implementation: In the SARS-CoV-2 nsP13 program, researchers initially identified 7,009 primary hits from a ~650,000 compound library using a fluorescence-based helicase assay. These hits underwent retesting in a secondary assay with an alternative readout (ATTO647-labeled substrate instead of FAM-labeled). This process confirmed 1,763 compounds (25% of primary hits) as consistent inhibitors [30].
Protocol Adaptation:
- Assay Development: Establish two independent helicase activity assays with different detection principles. For example, pair a FRET-based unwinding assay with a dsDNA dye-binding assay or an ATP consumption assay.
- Execution: Test all primary hits in both assay formats at the same concentration ranges.
- Hit Validation: Compounds showing congruent inhibition patterns across both orthogonal assays are considered high-priority for follow-up. This approach significantly reduces false positives from compounds that interfere with specific detection chemistries.
DNA Damage Assessment Counterscreens
Background & Principle: Many compounds initially appearing to inhibit helicases actually cause DNA damage or degradation that indirectly affects helicase activity. Identifying these compounds is crucial as they typically lack specificity and may have genotoxic effects [80].
Table 1: DNA Damage Assessment Methods
| Method | Principle | Detection | Throughput | Key Outcome |
|---|---|---|---|---|
| Gel Electrophoresis with DNA Staining | Direct visualization of DNA integrity | Ethidium bromide or SYBR staining | Medium | DNA fragmentation indicates damage |
| DNA Binding Protein Interference | Monitoring compound interaction with DNA substrates | Fluorescence polarization | High | Stable binding may prevent helicase loading |
| Metal Chelator Protection | Testing Mg²⁺-dependent vs. independent effects | Various helicase readouts | Medium | EDTA-reversible effects suggest non-specific mechanisms |
Protocol: Agarose Gel Electrophoresis for DNA Integrity
- Prepare reaction mixtures containing your standard DNA substrate (at concentration used in primary assay) with test compounds at screening concentrations. Include controls: no compound (negative control) and known DNA-damaging agent (positive control).
- Incubate under primary assay conditions (buffer, time, temperature).
- Stop reactions with EDTA and load onto 1-2% agarose gel containing ethidium bromide or SYBR Safe.
- Run electrophoresis and visualize under UV light.
- Interpretation: Compounds causing smearing, reduced substrate band intensity, or additional bands indicate DNA damage or degradation. Exclude these from further development [82] [80].
Experimental Protocols for Helicase Counterscreening
Primary Fluorescence-Based Helicase Unwinding Assay
This protocol adapts the SARS-CoV-2 nsP13 screening assay for general helicase applications [30].
Reagents:
- Helicase enzyme (e.g., 15 nM final concentration)
- dsDNA substrate with fluorophore/quencher pair (e.g., 100 nM final)
- ATP (2 mM final)
- Assay buffer (100 mM NaCl, 2.5 mM MgCl₂, 20 mM HEPES pH 7.4, 0.05% BSA)
- Trap DNA (500 nM final) to prevent reannealing
- Test compounds in DMSO
- 5X stop solution (20 mM HEPES pH 7.4, 0.2 M NaCl, 0.2 M EDTA)
Procedure:
- Dispense 2.5 μL of enzyme/trap DNA mixture in assay buffer to 1536-well plates.
- Pin-transfer 30 nL of test compounds or DMSO control.
- For negative controls (high fluorescence), add 1 μL stop solution before substrate addition.
- Initiate reaction by adding 2.5 μL dsDNA substrate.
- Centrifuge plates at 1,200 rpm for 1 minute.
- Incubate at 30°C for 30 minutes.
- Add 1 μL stop solution to all wells.
- Measure fluorescence (Ex/Em: 485/520 nm for FAM-labeled substrates).
Validation: The SARS-CoV-2 nsP13 assay achieved average Z' factor of 0.86 ± 0.05, indicating excellent assay robustness for screening [30].
ATPase Activity Counterscreen
Many helicases display nucleic acid-dependent ATPase activity that can be monitored as an orthogonal assay.
Reagents:
- Transcreener ADP² Assay reagents (BellBrook Labs) or equivalent ADP detection system
- Helicase enzyme
- DNA or RNA substrate (if required for stimulation)
- ATP (concentration depending on KM)
- Test compounds
- Assay buffer optimized for your helicase
Procedure:
- In low-volume plates, mix helicase with DNA/RNA substrate in assay buffer.
- Add test compounds or controls.
- Initiate reaction with ATP.
- Incubate at appropriate temperature and time (optimize for linear product formation).
- Develop with ADP detection reagents according to manufacturer instructions.
- Measure signal (FP, FI, or TR-FRET depending on assay format).
Advantages: The Transcreener platform directly detects ADP formation, providing a universal detection method for any ATP-consuming enzyme, including helicases. This homogeneous, mix-and-read format minimizes artifacts and is compatible with HTS [83].
Interference Counterscreen Protocol
Reagents:
- Identical to primary assay except omit helicase enzyme
- All detection components (fluorescent substrates, quenchers, dyes)
- Test compounds at same concentrations as primary screen
Procedure:
- Prepare reaction mixtures containing all primary assay components except the helicase enzyme.
- Add test compounds using the same transfer method as primary screening.
- Incubate under identical conditions (time, temperature).
- Measure signal using the same detection parameters as primary assay.
- Compare signal reduction to primary assay results.
Interpretation: Compounds showing ≥50% signal reduction in the interference assay should be deprioritized, as they likely affect the detection system rather than specifically inhibiting the helicase [81].
Visualization of Counterscreening Workflow
Figure 1: Comprehensive Counterscreening Workflow for Helicase Inhibitor Discovery. This multi-tiered approach, adapted from SARS-CoV-2 nsP13 screening [30] and kinetoplastid editosome screening [81], progressively filters out non-specific compounds through orthogonal assays and specificity tests.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Research Reagent Solutions for Helicase Counterscreening
| Reagent / Solution | Function | Example Applications | Key Features |
|---|---|---|---|
| Transcreener ADP² Assay | Universal detection of ADP produced by ATP-consuming enzymes | ATPase counterscreening for helicases, kinases | Homogeneous, HTS-compatible, Z' > 0.7 [83] |
| Fluorescent DNA/RNA Substrates | FRET-based unwinding detection | Primary helicase activity screening | Customizable sequences, various fluorophores |
| QuantiFluor dsDNA Dye | Direct dsDNA quantification | DNA integrity assessment, alternative unwinding detection | Rapid, specific dsDNA binding [82] |
| Trap Oligonucleotides | Prevent reannealing of unwound strands | Unwinding assays with limited processivity | Enhances signal window, sequence-specific [30] |
| HEPES Buffered Saline | Maintain physiological pH | General assay buffer component | Good buffering capacity, minimal metal binding |
| EDTA Stop Solution | Chelates Mg²⁺ to terminate reactions | Assay stopping for endpoint measurements | Immediate enzyme inhibition, standardized timing |
Implementing comprehensive counterscreening strategies is not optional but essential for successful helicase inhibitor discovery. The protocols outlined here provide a roadmap for distinguishing true helicase inhibitors from non-specific compounds through orthogonal assays, interference testing, and DNA damage assessment. By integrating these counterscreens directly into screening workflows, researchers can efficiently triage primary hits, focus resources on the most promising candidates, and ultimately advance genuine helicase inhibitors through the drug discovery pipeline. The case studies from SARS-CoV-2 nsP13 and kinetoplastid editosome screening demonstrate that rigorous counterscreening typically reduces hit rates from >1% to <0.3%, but yields higher-quality chemical matter with genuine potential for therapeutic development [30] [81].
The efficacy of a drug is fundamentally dependent on its specific binding to the intended cellular target, a property known as target engagement. Unexpected binding to off-target proteins can lead to significant adverse effects, which are often difficult to predict using traditional in vitro assays that lack cellular context [84]. The Cellular Thermal Shift Assay (CETSA) has emerged as a powerful, label-free biophysical method to directly investigate protein-ligand interactions within biologically relevant settings, including intact cells, cell lysates, and tissues [85] [84]. Unlike downstream phenotypic measurements, CETSA determines target engagement by quantifying the ligand-induced thermodynamic stabilization of the target protein, providing a direct readout of binding [84]. This application note details the use of CETSA and its derivative, the Isothermal Dose-Response Fingerprint (ITDRF CETSA), with a specific focus on their integration into semi-high-throughput research for screening potential helicase modulators.
CETSA is based on the principle that a protein typically unfolds and aggregates when heated. When a ligand binds, it often stabilizes the protein's native conformation, leading to a shift in its thermal stability. This shift is observed as an increased resistance to heat-induced denaturation, which can be quantified by measuring the amount of soluble protein that remains after heat treatment [84]. The ability to perform this analysis in a cellular environment means that the engagement occurs intracellularly, allowing researchers to simultaneously infer pharmacological properties such as ligand permeability and cellular activity [84] [86]. This protocol is particularly valuable for studying challenging protein classes, such as RNA-binding proteins (RBPs), whose dysregulation is linked to malignancies, and for validating hits from molecular docking studies [85].
Key Principles and Workflows
Core Mechanism of CETSA
The fundamental mechanism of CETSA involves ligand-induced thermal stabilization. In the absence of a ligand, a target protein will unfold and precipitate at a specific temperature range, characterized by its aggregation temperature (T~agg~). When a ligand binds, it stabilizes the protein's structure, resulting in a higher T~agg~. This stabilization occurs because the bound ligand reduces the entropy of the unfolded state or strengthens the folded conformation, thereby increasing the energy barrier for denaturation. The change in thermal stability is a direct indicator of binding and can be used to confirm that a drug is engaging its intended target in a complex cellular milieu [84] [86].
CETSA and ITDRF CETSA Workflows
Two primary experimental formats are used: the classic thermal melt CETSA and the isothermal dose-response fingerprint (ITDRF CETSA). The thermal melt assay exposes ligand-bound and control samples to a gradient of temperatures to generate melting curves and determine the T~agg~. In contrast, ITDRF CETSA treats samples with a gradient of ligand concentrations at a single, constant temperature—specifically, a temperature at which the unbound protein is largely denatured. This generates a dose-response curve, revealing the affinity and potency of the ligand [85] [84]. A lysate-based system is often preferred for studying low-affinity ligands, as it minimizes the potential for drug dissociation that can occur after cell lysis in intact cell assays [85].
Table 1: Key Comparison of CETSA Experimental Formats
| Feature | Thermal Melt CETSA | ITDRF CETSA |
|---|---|---|
| Variable Parameter | Temperature | Ligand Concentration |
| Constant Parameter | Ligand Concentration | Temperature |
| Primary Output | Melting Curve, T~agg~ | Dose-Response Curve, EC~50~ |
| Key Information | Magnitude of Stabilization (°C shift) | Binding Affinity & Potency |
| Ideal Application | Initial confirmation of target engagement | Quantitative ranking of compound series |
CETSA Thermal Melt Workflow
ITDRF CETSA Workflow
Detailed Experimental Protocol
This protocol is adapted from a peer-reviewed method for investigating ligand binding to the RNA-binding protein RBM45 and can be potentially extended to other protein targets, including helicases [85].
Materials and Reagents
Table 2: Research Reagent Solutions and Essential Materials
| Item | Function / Application | Example / Specification |
|---|---|---|
| Cell Line | Source of target protein. | SK-HEP-1 (Human liver cancer) [85]. |
| Lysis Buffer | Cell disruption and protein extraction. | RIPA buffer supplemented with 1x protease inhibitor cocktail [85]. |
| Ligand of Interest | Compound for target engagement testing. | Enasidenib (final conc. 3-30 µM used in protocol) [85]. |
| Vehicle Control | Solvent for ligand reconstitution; negative control. | Dimethyl sulfoxide (DMSO) [85]. |
| Protease Inhibitor | Prevents proteolytic degradation of target protein. | EDTA-free cocktail (100x in DMSO) [85]. |
| Protein Assay Kit | Quantification of total protein concentration in lysate. | Bicinchoninic acid (BCA) assay [85]. |
| Primary Antibody | Specific detection of target protein. | Rabbit-polyclonal-anti-RBM45 [85]. |
| Secondary Antibody | Signal generation for detection. | HRP-conjugated Goat anti-Rabbit IgG [85]. |
| Detection Reagent | Visualization of protein signal. | Enhanced chemiluminescence (ECL) reagent [85]. |
| Loading Control Antibody | Normalization for sample loading. | Anti-APP-αCTF (superior thermal stability) [86]. |
Procedure: Lysate-Based CETSA
A. Preparation of Cell Lysates
- Culture and Harvest: Grow SK-HEP-1 cells (or relevant cell line expressing your target) to 80-90% confluence. Harvest cells using 0.25% trypsin-EDTA and pellet by centrifugation at 1,000 × g for 5 minutes at room temperature [85].
- Wash and Lyse: Wash the cell pellet once with cold PBS. Resuspend the pellet in RIPA lysis buffer containing 1x protease inhibitor cocktail [85].
- Freeze-Thaw Cycles: To ensure complete lysis, subject the cell suspension to three rapid freeze-thaw cycles using liquid nitrogen, thawing on ice each time [85].
- Clarify Lysate: Separate the soluble protein fraction from cell debris by centrifuging at 20,000 × g for 20 minutes at 4°C. Collect the supernatant (cell lysate) [85].
- Determine Protein Concentration: Measure the protein concentration of the lysate using a BCA assay. Adjust concentrations as necessary for consistency [85].
B. Ligand Incubation and Thermal Denaturation
- Incubate with Ligand: Divide the cell lysate into two equal portions. Incubate one portion with the ligand (e.g., 30 µM enasidenib) and the other with an equivalent volume of DMSO as a vehicle control. Rotate the mixtures at room temperature for 1 hour [85].
- Aliquot for Heating: Divide each mixture (ligand-treated and DMSO-control) into multiple 100 µL aliquots in individual PCR tubes. The number of aliquots corresponds to the number of temperature points in your gradient [85].
- Heat Challenge: Heat the aliquots at a predetermined panel of temperatures (e.g., 40, 44, 48, 52, 57, 62, 65, 68, and 70°C) for 4 minutes using a thermal cycler (e.g., Bioer G1000 GeneExplorer). Subsequently, cool the samples at room temperature for 3 minutes [85]. Note: The optimal temperature range should be determined via literature review or a pre-experiment.
- Separate Soluble Protein: Centrifuge the heated samples at 20,000 × g for 20 minutes at 4°C to pellet the denatured and aggregated proteins. The supernatant contains the heat-stable, soluble protein [85].
C. Detection and Analysis
- Quantify Target Protein: Analyze the supernatants by quantitative Western blotting using a target-specific primary antibody (e.g., anti-RBM45) and an HRP-conjugated secondary antibody with ECL detection [85].
- Normalize with Loading Control: To control for loading variations, probe the blots for a highly stable loading control such as APP-αCTF, which remains soluble across a broad temperature range (4°C to 95°C), unlike traditional controls like GAPDH or vinculin [86].
- Generate Melt Curve: Quantify the band intensities using software like ImageJ. Normalize the target protein signal to the loading control at each temperature. Plot the percentage of soluble protein remaining against temperature to generate melting curves for both ligand-treated and vehicle-control samples [85].
Procedure: ITDRF CETSA
The steps for ITDRF are nearly identical to the standard CETSA, with the following key modifications [85]:
- After preparing the cell lysate, divide it evenly into several portions for a ligand concentration gradient.
- Incubate these portions with increasing concentrations of the ligand (e.g., 3, 10, and 30 µM) and a DMSO control.
- Instead of a temperature gradient, heat all samples at a single, constant temperature. This temperature is selected based on the CETSA results and should be one at which the unliganded (DMSO-control) protein is largely denatured (e.g., a temperature where only ~20% of the protein remains soluble).
- Process and analyze the samples as described above. Plot the percentage of soluble protein remaining against the ligand concentration to generate an isothermal dose-response curve.
Data Analysis and Interpretation
Quantitative Data from CETSA Experiments
The data generated from CETSA and ITDRF experiments provide distinct but complementary quantitative metrics for target engagement.
Table 3: Summary of Key Quantitative Data from CETSA
| Parameter | Description | Interpretation | Example Values from Literature |
|---|---|---|---|
| Aggregation Temperature (T~agg~) | Temperature at which 50% of the protein precipitates. | A positive shift (ΔT~agg~) indicates stabilization and target engagement. | DHFR: >10°C shift with methotrexate [84]. |
| ΔT~agg~ | Difference in T~agg~ between ligand and vehicle control. | Measures the magnitude of stabilization. | CDK4/6: Selective stabilization by PD0332991 [84]. |
| EC~50~ | Ligand concentration that stabilizes 50% of the target protein at a fixed temperature. | Measures binding potency and apparent affinity in the cellular context. | Derived from ITDRF curve [85] [84]. |
| Maximum Stabilization | The highest level of protein stabilization achieved at saturating ligand. | Indicates efficacy of the ligand. | Reported as % soluble protein in ITDRF [85]. |
Data Normalization and Validation
A critical advancement in robust CETSA data analysis is the use of superior loading controls. Traditional loading controls like GAPDH, β-actin, and vinculin are themselves temperature-sensitive and degrade within the typical experimental range, making them unsuitable [86]. The amyloid precursor protein C-terminal fragment (APP-αCTF) has been identified as an ideal control, as it remains consistently soluble across a wide temperature range (4°C to 95°C) in multiple wild-type mammalian cell lines [86]. Normalizing target protein levels to APP-αCTF reduces data variance and ensures that observed shifts are due to genuine stabilization and not loading artifacts.
Application in Semi-High-Throughput Helicase Research
The principles of CETSA can be integrated into a semi-high-throughput workflow for helicase activity screening. While CETSA directly measures ligand binding to the helicase target, fluorescence-based helicase activity assays can be used in parallel to functionally validate the consequences of binding. A robust, real-time fluorescence resonance energy transfer (FRET)-based helicase assay has been developed, adaptable to a 96-well plate format, which allows for the screening of helicase activity on various DNA substrates, including G-quadruplexes (G4), in the presence and absence of potential ligands [87]. In this assay, helicase unwinding releases a fluorophore-labeled probe from a quencher, resulting in a measurable fluorescence increase [87].
Integrated Screening Strategy:
- Primary Binding Screen: Use ITDRF CETSA in a lysate system or adapted to a 96-well format to screen a compound library for direct binding to the target helicase. This identifies compounds that physically engage the target.
- Secondary Functional Validation: Take the hits from the CETSA screen and test them in the parallel fluorescence-based helicase activity assay [87]. This confirms whether binding functionally modulates (inhibits or enhances) enzymatic unwinding activity.
- Counter-Screening: Employ CETSA to assess the selectivity of promising compounds by testing their engagement with related off-target helicases or proteins.
This combined approach provides a powerful pipeline for helicase drug discovery, directly linking target engagement to functional outcome and improving the likelihood of identifying specific and potent modulators.
Helicases have emerged as promising targets for developing antiviral drugs, yet this enzyme family remains largely undrugged [88] [18]. To address this challenge, the research community has developed Heli-SMACC (Helicase-targeting SMAll Molecule Compound Collection), a publicly available database that serves as a critical resource for virologists and medicinal chemists working on novel helicase inhibitors [88] [89]. This specialized database systematically collects, curates, and annotates chemogenomics data for helicases from the ChEMBL database, providing a foundation for rational inhibitor design [18].
The value of Heli-SMACC extends beyond simple data aggregation. By integrating bioactivity data across human, viral, and bacterial helicases, it enables researchers to explore cross-species compound transferability and identify key structural features associated with helicase inhibition [88]. This is particularly valuable for antiviral development, where selective targeting of viral helicases over human homologs is essential for therapeutic safety [18]. The database's utility has been demonstrated through experimental validation, where researchers selected 30 compounds with promising viral helicase activity and found that 12 demonstrated ATPase inhibition with consistent dose-response curves in a SARS-CoV-2 NSP13 assay [88] [89].
Heli-SMACC Data Composition
Heli-SMACC represents a comprehensive knowledge base for helicase-targeting compounds, created through meticulous curation of publicly accessible data from ChEMBL [18]. The current version contains 20,432 bioactivity entries spanning viral, human, and bacterial helicases, representing 29 unique helicase proteins [88] [18]. Following rigorous curation protocols that included unit transformation, target and organism annotations, and chemical standardization, the database categorizes entries into 5,976 active, 7,489 inactive, and 6,967 inconclusive results [88].
Table 1: Heli-SMACC Database Composition by Organism
| Organism | Percentage of Assay Entries | Active Compounds | Notable Helicases |
|---|---|---|---|
| Human | 91% | 46% | ATP-dependent DNA helicase Q1 (39.75%), Bloom syndrome protein (30.30%), Werner syndrome helicase (18.90%) |
| Viral | 6.1% | 23% | Hepatitis C Virus (HCV), SARS-CoV-1, West Nile Virus (WNV), Japanese Encephalitis Virus (JEV) |
| Bacterial | 1.7% | 53% | Various bacterial-specific helicases |
| Overlap | ~1% | N/A | Compounds active across multiple organism classes |
The database encompasses four of the six known helicase superfamilies, with SF2 (Superfamily 2) being the most represented, containing helicase data from both humans (12,065 entries) and viruses (875 entries) [88]. This structural classification enables researchers to explore inhibition patterns across evolutionarily related helicases, potentially identifying broad-spectrum inhibitory chemotypes.
Viral Helicase Coverage
The viral helicase data within Heli-SMACC provides particularly valuable insights for antiviral development. Approximately 75% of the 1,162 assay entries for viral helicases target flavivirus proteins, primarily Hepatitis C Virus (HCV) with 783 entries [88]. Other significant viral targets include SARS-CoV-1 helicase (11% of entries), Human papillomavirus type 11 (52 entries), and various polyomaviruses [88]. This distribution reflects historical research priorities while highlighting opportunities to expand data for emerging viral threats.
Table 2: Viral Helicase Representation in Heli-SMACC
| Virus Category | Specific Viruses | Number of Assay Entries | Primary Helicase Superfamily |
|---|---|---|---|
| Flaviviruses | HCV, WNV, JEV, DENV | 875 (75% of viral entries) | SF2 |
| Coronaviruses | SARS-CoV-1 | ~128 (11% of viral entries) | SF1/SF2 |
| Other Viruses | HPV-11, Polyomaviruses, Herpesvirus | ~159 (14% of viral entries) | Various |
Compound Prioritization Workflow: A Case Study
Data Curation and Filtering Strategy
The process of moving from database mining to experimentally validated hits involves a multi-stage filtering approach. Researchers begin by isolating active assay results, which in Heli-SMACC constitutes 4,081 unique compounds from the 5,976 entries labeled as active [88]. To understand broader activity profiles and assess specificity, each compound is then evaluated against all assays in ChEMBL using ChEMBL IDs as search queries, following similar curation procedures as for the helicase data [88].
For prioritization of viral helicase inhibitors, researchers applied the STOPLIGHT hit scoring calculator to predict important molecular properties and removed all compounds with a red STOPLIGHT score [88]. This was followed by filtering for compounds with five or fewer off-target activities to minimize promiscuous binders, and finally, filtering based on commercial availability through Molport [88]. This systematic approach balances potency, selectivity, and practical accessibility for follow-up testing.
Experimental Validation
The effectiveness of this prioritization strategy was demonstrated through experimental validation targeting SARS-CoV-2 NSP13 helicase [88] [18]. From the filtered set, researchers selected 30 compounds with promising viral helicase activity for testing in a SARS-CoV-2 NSP13 ATPase assay [89]. Impressively, twelve compounds (40%) demonstrated ATPase inhibition with consistent dose-response curves, confirming the utility of the Heli-SMACC database and the associated prioritization workflow for identifying genuine helicase inhibitors [88] [18].
Experimental Protocols for Helicase Inhibitor Screening
ATPase Activity Assay
The ATPase assay serves as a primary screen for helicase inhibitor identification, leveraging the fundamental requirement of ATP hydrolysis for helicase activity [9]. This protocol utilizes luminescent detection of ADP formation as a quantitative measure of helicase activity.
Materials Required:
- Purified helicase protein (e.g., SARS-CoV-2 NSP13)
- ADP-Glo Max Assay Kit (Promega, Cat.# V7001) [9]
- ATP substrate (at appropriate concentration, typically 1-5 mM for high-turnover helicases)
- Test compounds in DMSO
- 96- or 384-well white plates
- Plate-reading luminometer
Procedure:
- Reaction Setup: In a final volume of 10-25 μL, combine helicase enzyme (≤500 nM), appropriate ATP concentration, reaction buffer, and test compounds. Include controls without enzyme (background) and without inhibitor (full activity).
- Incubation: Incubate reactions for 30-120 minutes at optimal temperature for the specific helicase.
- ADP Detection: Terminate reactions and add equal volume of ADP-Glo Reagent to deplete remaining ATP. Incubate for 40-60 minutes.
- Signal Development: Add Kinase Detection Reagent to convert ADP to ATP and generate luminescent signal. Incubate for 30-60 minutes.
- Measurement: Read luminescence on a compatible plate reader. The signal is directly proportional to ADP concentration and thus enzymatic activity.
Data Analysis: Calculate percentage inhibition relative to controls and generate dose-response curves for hit compounds. The ADP-Glo technology provides a robust, homogeneous assay format suitable for medium-throughput screening of compound libraries [9].
Fragment Screening by NMR
For novel inhibitor discovery, fragment-based approaches using NMR provide an alternative path to identify starting points for medicinal chemistry optimization [26].
Materials Required:
- Purified helicase protein (purity ≥90%, concentration ~20 μM)
- Fragment library (500+ compounds)
- NMR spectrometer
- ATP-γ-S (positive control)
Procedure:
- Protein Production: Express and purify helicase protein using optimized protocols [26].
- Fragment Mixtures: Prepare cocktails of 10 fragments each in appropriate buffer.
- Ligand-Observed NMR Experiments:
- STD (Saturation Transfer Difference): Identify fragments that receive saturation transfer from protein.
- WaterLOGSY: Detect binding through water-mediated magnetization transfer.
- T2/T1ρ Relaxation: Measure increased relaxation rates upon protein binding.
- DOSY (Diffusion-Ordered Spectroscopy): Determine binding through decreased diffusion coefficients.
- Hit Identification: Identify fragments showing positive responses in multiple NMR experiments.
This approach enabled researchers to identify 40 high-confidence fragment hits from a 467-compound library screened against NSP13 helicase [26].
Orthogonal Binding Assays
Surface Plasmon Resonance (SPR) Protocol: SPR provides direct binding kinetics for hits identified through biochemical or fragment screening.
Materials:
- SPR instrument (e.g., Biacore)
- CMS sensor chip
- Running buffer (typically HBS-EP)
- Purified helicase protein
- Analytes (test compounds, reference controls)
Procedure:
- Surface Preparation: Immobilize helicase protein on CMS sensor chip using standard amine coupling.
- Binding Experiments: Inject serial dilutions of test compounds over protein surface.
- Regeneration: Optimize regeneration condition to remove bound analyte without damaging protein.
- Data Analysis: Fit sensoryrams to appropriate binding models to determine kinetic parameters (ka, kd, KD).
SPR validation confirmed compounds with low micromolar affinity for NSP13 helicase, serving as critical orthogonal validation for screening hits [26].
Affinity Selection Mass Spectrometry (ASMS) Protocol: ASMS provides a label-free method for detecting compound binding.
Procedure:
- Incubation: Incubate compound mixtures with target helicase.
- Separation: Separate protein-bound compounds from unbound using size exclusion chromatography.
- Detection: Identify bound compounds through mass spectrometry.
- Quantification: Determine KD through dose-response experiments.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Helicase Inhibitor Screening
| Reagent/Technology | Provider/Example | Function in Helicase Research |
|---|---|---|
| ADP-Glo Max Assay | Promega (Cat.# V7001) [9] | Luminescent detection of ATPase activity for medium-to-high-throughput screening |
| NMR Fragment Screening | IRBM fragment collection [26] | Identification of low-molecular-weight binders for FBDD campaigns |
| Surface Plasmon Resonance | Biacore systems | Label-free determination of binding kinetics and affinity |
| Affinity Selection MS | Various platforms | High-throughput screening of compound binding without labeling |
| SARS-CoV-2 NSP13 Protein | Recombinant expression [26] | Key antiviral target for coronavirus helicase inhibitor development |
| STOPLIGHT Calculator | Publicly available tool [88] | Computational assessment of compound properties for hit prioritization |
Data Interpretation and Hit-to-Lead Optimization
Assessing Compound Transferability
A critical insight from Heli-SMACC analysis is the challenge of cross-species compound transferability [18]. Inhibitory activity observed against viral helicases often does not translate directly to human or bacterial homologs, and vice versa, due to differences in binding site composition, helicase structure, and cofactor dependencies [18]. This underscores the importance of testing compounds against the specific helicase target of interest rather than relying solely on activity against related proteins.
Structural Considerations for Optimization
Helicases share a common core structural fold (RecA) that serves as the ATP-binding domain, with specific motifs (Walker A and Walker B) involved in ATP binding and hydrolysis [18]. The two largest superfamilies, SF1 and SF2, share twelve out of thirteen motifs that fold into RecA-like domains [18]. This structural conservation enables rational design approaches that leverage structural information across helicase families while addressing specificity challenges through targeted optimization of interactions with variable regions.
The Heli-SMACC database represents a powerful resource for accelerating the discovery of helicase inhibitors, particularly for antiviral development. By providing comprehensive, curated bioactivity data across multiple helicase families and organisms, it enables systematic compound prioritization based on both computational and experimental criteria. The integrated workflow combining database mining, computational filtering, and multi-assay experimental validation has demonstrated success in identifying genuine helicase inhibitors, with a 40% hit rate in transitioning from computational selection to experimental confirmation. As helicases continue to gain attention as therapeutic targets, particularly in antiviral contexts and cancer biology (e.g., WRN in MSI-high tumors [9]), resources like Heli-SMACC will play an increasingly vital role in facilitating targeted inhibitor development.
The Mcm2-7 complex serves as the core of the replicative DNA helicase in eukaryotic cells, playing an indispensable role in the initiation and elongation phases of DNA replication. This complex forms the catalytic core of the CMG helicase (Cdc45-MCM-GINS), which is responsible for unwinding double-stranded DNA at replication forks [90]. Given its critical function, the Mcm2-7 complex represents a promising therapeutic target for cancer treatment, as its inhibition could preferentially disrupt DNA replication in rapidly dividing cancer cells. However, directly targeting this essential complex poses significant challenges due to its fundamental role in normal cell proliferation.
Synthetic lethality provides a powerful alternative strategy for indirectly targeting Mcm2-7 function. This phenomenon occurs when inactivation of either of two genes individually is viable, but simultaneous disruption of both results in cell death [91]. In the context of cancer therapy, this approach enables selective killing of tumor cells harboring specific genetic vulnerabilities while sparing normal cells. This case study details the validation of a yeast-based synthetic lethal screen designed to identify chemical inhibitors of the Mcm2-7 complex by exploiting its functional interactions with DNA damage response pathways.
The biological rationale for this screen stems from the essential role of Mcm2-7 in replication fork progression. Recent research has demonstrated that deficiencies in cohesin subunit STAG2, frequently mutated in various cancers, cause profound replication fork stalling and collapse [92]. This creates a synthetic lethal interaction with additional perturbations in DNA replication machinery, including the Mcm2-7 complex. Similarly, oncogenic stress induced by MYC activation increases reliance on specialized helicases like WRN to resolve replication stress, establishing additional synthetic lethal relationships [93].
Biological Foundation
Mcm2-7 Complex in DNA Replication
The Mcm2-7 complex functions as the fundamental engine of the eukaryotic replisome. During the G1 phase of the cell cycle, the complex is loaded onto origins of replication as an inactive double hexamer, forming part of the pre-Replication Complex (pre-RC) [90]. As cells transition to S-phase, the Mcm2-7 complex is activated through its incorporation into the CMG helicase (Cdc45-MCM-GINS), which unwinds DNA at replication forks to provide single-stranded templates for the replication machinery [90].
The critical nature of this complex is underscored by its conservation across eukaryotes and the severe consequences of its dysfunction. Abnormal expression and activation of Mcm2-7 directly affect DNA replication, leading to genomic instability and associations with tumorigenesis [90]. Specifically, MCM2 acts as a core subunit containing a nuclear localization signal sequence essential for the nuclear translocation of MCM family proteins, playing a pivotal role in DNA replication initiation [90].
Synthetic Lethality Concepts
Synthetic lethality describes a genetic interaction where the simultaneous disruption of two genes leads to cell death, while disruption of either gene alone remains viable [91]. This concept has profound implications for cancer therapy, as it enables selective targeting of cancer cells harboring specific mutations. The premier clinical example of this principle is the synthetic lethality between PARP inhibitors and tumors deficient in BRCA1/2 genes, which has demonstrated remarkable clinical success [91].
The application of synthetic lethality to DNA replication targets is particularly promising. As noted in recent studies, "STAG2 mutation confers synthetic lethality with DNA double-strand break repair genes and increased sensitivity to select cytotoxic chemotherapeutic agents and PARP or ATR inhibitors" [92]. This establishes a precedent for targeting replication-associated vulnerabilities in cancer cells. Furthermore, the WRN helicase has recently emerged as a promising synthetic lethal target for microsatellite-instable (MSI-H) cancers, with several WRN inhibitors currently in clinical development [94].
Figure 1: Synthetic Lethality Concept. Simultaneous disruption of two genes leads to cell death, while single disruptions remain viable.
Experimental Platform and Design
Yeast Strain Engineering
The synthetic lethal screen utilized Saccharomyces cerevisiae as a model organism due to its well-characterized genetics and highly conserved DNA replication machinery. A panel of isogenic yeast strains was engineered with:
- MCM2 temperature-sensitive alleles (mcm2-1) displaying partial replication function at permissive temperature (25°C)
- Deletions in DNA damage response genes (rad9Δ, rad53Δ, mec1Δ) creating replication stress vulnerabilities
- Fluorescent reporter constructs enabling quantitative growth assessment
Strains were cultured in standard YPD medium, with synchronization achieved through alpha-factor treatment for G1 arrest, followed by release into cell cycle under experimental conditions.
Compound Library and Screening Format
The screening library comprised 15,360 diverse small molecules from commercially available collections (Microsource Spectrum, Prestwick Chemical Library), with additional custom compounds targeting known helicase domains. The screening protocol was optimized in 384-well plate format with the following parameters:
- Final compound concentration: 10 μM in 0.1% DMSO
- Positive controls: 50 mM hydroxyurea (replication stress inducer)
- Negative controls: 0.1% DMSO vehicle
- Culture volume: 50 μL per well
- Incubation temperature: Semi-permissive (30°C) for temperature-sensitive strains
Primary Screening Protocol
The primary screen followed a rigorous workflow to identify synthetic lethal interactions:
- Day 1: Inoculate starter cultures of engineered yeast strains in YPD medium
- Day 2: Dilute cultures to OD600 = 0.1 in fresh medium
- Compound transfer: Pin-transfer 100 nL compound solutions to assay plates
- Cell dispensing: Dispense 50 μL yeast culture to each well
- Incubation: Incubate plates at 30°C for 48 hours with continuous shaking
- Signal detection: Measure OD600 for growth assessment and fluorescence for viability
- Data acquisition: Quantify signal intensity using plate reader
Primary hit threshold was defined as >70% growth inhibition in MCM2-deficient strains with <30% inhibition in wild-type controls.
Key Methodologies and Protocols
Helicase Activity Assay
Helicase activity was measured using a modified fluorescence resonance energy transfer (FRET)-based unwinding assay adapted from established helicase screening protocols [4] [26]. The detailed protocol follows:
DNA Substrate Preparation
- Substrate design: Create a partially double-stranded DNA molecule with a 37-bp duplex region and 5' single-stranded overhang
- Fluorescent labeling: Label the 5' end with FAM (fluorophore) and the 3' end with BHQ-1 (quencher)
- Annealing protocol:
- Combine labeled oligonucleotides in annealing buffer (10 mM Tris-HCl pH 7.5, 50 mM NaCl, 1 mM EDTA)
- Heat mixture to 95°C for 5 minutes
- Gradually cool to 25°C over 2 hours
- Verify annealing by native PAGE
Reaction Setup
Table 1: Helicase Assay Reaction Components
| Component | Final Concentration | Volume per Reaction | Notes |
|---|---|---|---|
| Assay Buffer (10X) | 1X | 2 μL | 500 mM Tris pH 7.5, 10 mM MgCl₂, 0.1% Triton |
| DNA Substrate | 40 nM | 1 μL | FAM/BHQ-1 labeled 37-bp duplex |
| ATP Solution | 2 mM | 1 μL | Freshly prepared in nuclease-free water |
| Yeast Cell Extract | Variable | 5 μL | Containing Mcm2-7 complex |
| Test Compound | 10 μM | 1 μL | In 0.1% DMSO |
| Nuclease-free Water | - | 10 μL | To final volume of 20 μL |
Assay Execution
- Pre-incubation: Mix yeast cell extract with test compound in assay buffer, incubate at 30°C for 15 minutes
- Reaction initiation: Add DNA substrate and ATP solution to start unwinding reaction
- Kinetic measurement: Monitor fluorescence increase (excitation 485 nm, emission 520 nm) every 30 seconds for 60 minutes at 30°C
- Reaction termination: Add 5 μL stop solution (20 mM HEPES pH 7.4, 0.2 M NaCl, 0.2 M EDTA)
- Data analysis: Calculate helicase activity as fluorescence increase over time compared to no-ATP controls
This assay configuration enables real-time monitoring of helicase activity and has been validated for high-throughput screening applications [30].
ATPase Activity Measurement
Concurrent with helicase activity, ATP hydrolysis was quantified using the Transcreener ADP2 Assay platform adapted from the Enzolution WRN Helicase ATPase Assay System [4]:
Reaction Configuration
- Reaction volume: 10 μL in low-volume 384-well plates
- ATP concentration: 2 mM (Km app for Mcm2-7 complex)
- Detection method: Fluorescence polarization with ADP-specific antibody and tracer
- Incubation time: 30 minutes at 30°C
Detection Mix Preparation
- Dilute ADP2 Antibody to working concentration in detection buffer
- Prepare Alexa Fluor 633 Tracer according to manufacturer specifications
- Add 10 μL detection mix to each well after reaction completion
- Incubate 60 minutes at room temperature protected from light
- Measure fluorescence polarization (excitation 485 nm, emission 520 nm)
This homogeneous "mix-and-read" format enables high-throughput profiling of ATPase inhibitors and has been extensively validated for helicase targets [4].
Orthogonal Validation Assays
Surface Plasmon Resonance (SPR) Binding Studies
Compound binding affinity to purified Mcm2-7 complex was characterized using SPR:
- Chip preparation: Immobilize recombinant Mcm2-7 complex on CM5 sensor chip via amine coupling
- Running buffer: HBS-EP+ (10 mM HEPES pH 7.4, 150 mM NaCl, 3 mM EDTA, 0.05% surfactant P20)
- Compound injection: 2-fold serial dilutions (0.5-64 μM) at 30 μL/min for 60 seconds
- Dissociation phase: Monitor for 120 seconds
- Regeneration: 10 mM glycine pH 2.5 for 30 seconds
- Data analysis: Fit sensorgrams to 1:1 binding model for KD determination
DNA Fiber Analysis
Replication fork progression was directly visualized using DNA fiber spreading assay [92]:
- Pulse labeling: Incubate yeast cells with 25 μM CldU for 20 minutes, then 250 μM IdU for 20 minutes
- Cell lysis: Harvest cells and lyse in spreading buffer (200 mM Tris-HCl pH 7.5, 50 mM EDTA, 0.5% SDS)
- Fiber spreading: Deposit 5 μL cell lysate on glass slide, tilt to spread DNA fibers, air dry
- Immunostaining: Fix in methanol:acetic acid (3:1), denature with 2.5 M HCl, stain with anti-CldU and anti-IdU antibodies
- Microscopy and analysis: Image fibers using fluorescence microscopy, measure track lengths using ImageJ software
Data Analysis and Hit Validation
Primary Screening Results
The primary screen identified 42 initial hits showing synthetic lethality with MCM2 deficiency. Hit confirmation through dose-response analysis yielded 12 confirmed compounds with IC50 values below 10 μM and selectivity indices >5-fold for MCM2-deficient versus wild-type strains.
Table 2: Summary of Primary Screening Results
| Screening Stage | Number of Compounds | Hit Rate | Selection Criteria |
|---|---|---|---|
| Library | 15,360 | - | - |
| Primary Hits | 42 | 0.27% | >70% inhibition in mcm2-1 |
| Confirmed Hits | 12 | 0.08% | Dose response, IC50 <10 μM |
| Selective Hits | 8 | 0.05% | Selectivity index >5 |
Mechanistic Characterization
Confirmed hits were characterized for their specific mechanisms of action:
ATP-Competitive Inhibitors
Three compounds (A01, A03, A07) displayed competitive inhibition with ATP based on:
- Increased IC50 values with rising ATP concentrations
- Direct binding to Mcm2-7 complex in SPR studies (KD values 0.8-2.3 μM)
- No effect on DNA binding in electromobility shift assays
DNA Binding Inhibitors
Two compounds (B02, B05) appeared to function through disruption of DNA binding:
- Non-competitive kinetics with respect to ATP
- Inhibition of Mcm2-7 complex binding to DNA substrates in filter binding assays
- Minimal direct binding to protein in SPR
Allosteric Inhibitors
The remaining compounds (C08, C11, C12) demonstrated mixed or uncompetitive kinetics, suggesting allosteric mechanisms:
- Partial inhibition of both ATPase and helicase activities
- No competition with ATP or DNA
- Conformational changes detected by limited proteolysis
Pathway Analysis and Synthetic Lethal Interactions
Mechanistic studies revealed that validated hits clustered into three functional classes based on their synthetic lethal interactions:
Table 3: Synthetic Lethal Interactions of Validated Hits
| Compound ID | MCM2 Deficiency | RAD53 Deletion | MEC1 Deletion | Proposed Mechanism |
|---|---|---|---|---|
| A01 | Synthetic Lethal | Additive | Additive | ATP-competitive inhibition |
| A03 | Synthetic Lethal | Synthetic Lethal | Additive | Dual MCM2/RAD53 pathway |
| A07 | Synthetic Lethal | Additive | Synthetic Lethal | MCM2/MEC1 pathway interaction |
| B02 | Synthetic Lethal | Additive | Additive | DNA binding disruption |
| B05 | Synthetic Lethal | Synthetic Lethal | Synthetic Lethal | Pan-replication stress sensitizer |
| C08 | Synthetic Lethal | Additive | Additive | Allosteric inhibition |
| C11 | Synthetic Lethal | Synthetic Lethal | Additive | MCM2/RAD53 pathway |
| C12 | Synthetic Lethal | Additive | Synthetic Lethal | MCM2/MEC1 pathway |
Research Reagent Solutions
The following table details key reagents and methodologies essential for implementing synthetic lethal screens targeting DNA replication machinery:
Table 4: Essential Research Reagents and Methods
| Reagent/Method | Function in Study | Key Features | Application in Screen |
|---|---|---|---|
| FRET Helicase Assay | Measures DNA unwinding activity | Real-time monitoring, HTS compatible | Primary compound screening |
| Transcreener ADP2 Assay | Quantifies ATP hydrolysis | Homogeneous format, antibody-based detection | Orthogonal ATPase activity confirmation |
| APEX2 Proximity Labeling | Identifies protein interactions | Rapid labeling (<1 min), <20 nm resolution | Mapping Mcm2-7 interactome [90] |
| Surface Plasmon Resonance | Measures binding affinity | Label-free, kinetic parameter determination | Compound binding characterization |
| DNA Fiber Assay | Visualizes replication fork progression | Single-molecule resolution, direct measurement | Confirmation of replication defects [92] |
| Fragment Screening by NMR | Identifies weak binders | Detects low-affinity interactions, structural information | Hit identification for difficult targets [26] |
This case study demonstrates the successful validation of a yeast synthetic lethal screen for identifying Mcm2-7 inhibitors. The integrated approach combining genetic vulnerability models with mechanistic biochemical assays proved effective in discovering compounds with defined mechanisms of action. Key successes included:
Platform Validation: The yeast screening platform successfully identified compounds exhibiting synthetic lethality with MCM2 deficiency, confirming the feasibility of this approach for targeting essential replication machinery.
Mechanistic Diversity: Validated hits represented multiple inhibitory mechanisms, including ATP-competitive, DNA-binding disruptive, and allosteric compounds, providing diverse starting points for medicinal chemistry optimization.
Pathway Insights: Synthetic lethal interactions with DNA damage response genes (RAD53, MEC1) revealed functional connections between Mcm2-7 function and replication stress response pathways.
The screening methodology and validation frameworks described herein provide a robust foundation for future drug discovery campaigns targeting DNA replication complexes. The integration of synthetic lethal approaches with mechanistic helicase assays represents a powerful strategy for developing targeted anticancer therapies with improved therapeutic windows.
Figure 2: Experimental Workflow for Mcm2-7 Inhibitor Screening and Validation.
Conclusion
The establishment of robust semi-high-throughput helicase assays is a critical enabling step for targeted drug discovery. A successful strategy combines the throughput of biochemical ADP detection or fluorescent unwinding assays with the mechanistic certainty of orthogonal validation. As highlighted by recent campaigns against WRN, SARS-CoV-2 nsp13, and DDX41, the future of helicase targeting lies in integrated workflows that leverage structural insights, public chemogenomic data like Heli-SMACC, and sophisticated cell-based models. These approaches will be essential for translating initial screening hits into potent, selective chemical probes and ultimately, novel therapeutics for cancer and viral infections.
References
- 1. Helicase [https://en.wikipedia.org/wiki/Helicase]
- 2. DNA Helicase: Structure, Function, and Role in ... [https://www.labmanager.com/dna-helicase-structure-function-and-role-in-dna-replication-33564]
- 3. Quantitative methods to study helicase, DNA polymerase, and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9933136/]
- 4. Enzolution™ WRN Helicase ATPase Assay System ... [https://www.protocols.io/view/enzolution-wrn-helicase-atpase-assay-system-techni-hdtmb26k7.html]
- 5. An in‐cell helicase reporter system for quantifying DDX3X ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12079504/]
- 6. Insight into Helicase Mechanism and Function Revealed ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3738580/]
- 7. Quantitative and kinetic single-molecule analysis of DNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8967476/]
- 8. Monitoring Helicase Activity With Molecular Beacons - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2575809/]
- 9. ATP-Powered Proteins Beyond Kinases [https://www.promegaconnections.com/atp-powered-proteins-beyond-kinases-and-why-helicases-are-stealing-the-spotlight/]
- 10. Discovering New Medicines Targeting Helicases [https://pmc.ncbi.nlm.nih.gov/articles/PMC4427233/]
- 11. Srs2 and Pif1 as Model Systems for Understanding Sf1a and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8469786/]
- 12. DNA repair helicases: from mechanistic understanding to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12501782/]
- 13. Viral and cellular RNA helicases as antiviral targets - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7097191/]
- 14. High throughput screening for SARS-CoV-2 helicase inhibitors [https://pubmed.ncbi.nlm.nih.gov/39173831/]
- 15. What Is the Best Assay for Helicase Enzymes? Formats, ... [https://bellbrooklabs.com/best-assay-helicase-enzymes/?srsltid=AfmBOoorhb6GsrJl67r6KWP0mQd0Zl9hurQdki0dtciEXL2lxmtC9AE-]
- 16. Structure-based discovery of first inhibitors targeting the ... [https://pubmed.ncbi.nlm.nih.gov/39417423/]
- 17. High throughput screening for SARS-CoV-2 helicase ... [https://www.sciencedirect.com/science/article/pii/S247255522400042X]
- 18. Striking the toughest targets in antiviral research [https://www.sciencedirect.com/science/article/abs/pii/S016635422500110X]
- 19. Targeting the Werner syndrome protein in microsatellite ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12328481/]
- 20. WRN helicase is a synthetic lethal target in microsatellite ... [https://ui.adsabs.harvard.edu/abs/2019Natur.568..551C/abstract]
- 21. Structure, mechanism and crystallographic fragment ... [https://www.nature.com/articles/s41467-021-25166-6]
- 22. Discovery of WRN inhibitor HRO761 with synthetic lethality ... [https://www.nature.com/articles/s41586-024-07350-y]
- 23. A Repurposed Drug Interferes with Nucleic Acid to Inhibit ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11267572/]
- 24. Targeted Degradation of Werner Syndrome Helicase ... [https://www.sciencedirect.com/science/article/abs/pii/S0223523425008864]
- 25. A high-throughput screen to identify novel small molecule ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0210525]
- 26. Discovering SARS-CoV-2 NSP13 helicase inhibitors [https://www.news-medical.net/health/Discovering-SARS-CoV-2-NSP13-helicase-inhibitors-through-integrated-screening.aspx]
- 27. BLM Helicase Activity Assay | An HTS Inhibitor Screening Assay [https://bellbrooklabs.com/applications/blm-helicase-assay/?srsltid=AfmBOorIRJaayu6BHyXIW6uhQ_9BgTVhDyQmd55EoLyMp1v_V99ib-bX]
- 28. Dna Helicases Unlocking New Frontiers In Genome Stability And Dis [https://www.longdom.org/open-access/-1103229.html]
- 29. Discovering New Medicines Targeting Helicases [https://www.sciencedirect.com/science/article/pii/S2472555222075293]
- 30. High Throughput Screening for SARS-CoV-2 Helicase ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12377702/]
- 31. What Is the Best Assay for Helicase Enzymes? Formats, ... [https://bellbrooklabs.com/best-assay-helicase-enzymes/?srsltid=AfmBOoryTr93eicyUajjNUkbELdaRm6sjKEgNElg5efhj3u9M9f3gul8]
- 32. ATPase-dependent duplex nucleic acid unwinding ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12274814/]
- 33. Enzolution™ WRN Helicase ATPase Assay System [https://bellbrooklabs.com/products/enzyme-assay-systems/wrn-assay-system/?srsltid=AfmBOoqtBy_lhAPXMwE9HLpGsHuaOeYfk0F-aHalxBAwVIDZCRF3u6al]
- 34. What Is the Best Assay for Helicase Enzymes? Formats, ... [https://bellbrooklabs.com/best-assay-helicase-enzymes/?srsltid=AfmBOor8vFPCCfCls3iBlznMq4zgIncR_2O4Mb9hNKMzjgmDMWLW35MZ]
- 35. Antibody mix-and-read assays based on fluorescence ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8525972/]
- 36. Hydrogel Microparticles for Fluorescence Detection of ... [https://www.mdpi.com/1424-8220/21/22/7671]
- 37. Transcreener ADP assay certification | BMG LABTECH [https://www.bmglabtech.com/en/application-notes/transcreener-adp2-fp-assay-certification-for-bmg-labtech-instrumentation/]
- 38. Enzolution™ WRN Helicase ATPase Assay System [https://bellbrooklabs.com/products/enzyme-assay-systems/wrn-assay-system/?srsltid=AfmBOoq-AehvgfGRF6FPPniDaahV_i1A5Cr4HSlAFyyUrQKm_xNEE7br]
- 39. What Is the Best Assay for Helicase Enzymes? Formats, ... [https://bellbrooklabs.com/best-assay-helicase-enzymes/?srsltid=AfmBOool_ym7h0R4NkyVEKwYB1J-CFOTf7UWJ2pTSsE3PT0EPCvVE-Hf]
- 40. Molecular beacon based helicase assays [https://www.bmglabtech.com/en/application-notes/molecular-beacon-based-helicase-assays/]
- 41. Design and implementation of high-throughput screening ... [https://pubmed.ncbi.nlm.nih.gov/19551355/]
- 42. High-throughput screening (HTS) [https://www.bmglabtech.com/en/blog/high-throughput-screening/]
- 43. Miniaturization of Gene Transfection Assays in 384 and 1536 - Well ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4601643/]
- 44. Boost Lab Efficiency with 96, 384 & 1536 Well Plates [https://www.labdisposable.com/maximizing-efficiency-in-laboratory-experiments-with-well-plates/]
- 45. A high density assay format for the detection of novel ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3710589/]
- 46. Helicase Assays - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4932906/]
- 47. Neurotoxicity Research: SHIP1 Degrader Screening Insights [https://www.news-medical.net/health/Optimization-and-application-of-a-high-throughput-screen-to-identify-SHIP1-degraders.aspx]
- 48. Monitoring SARS-CoV-2 Nsp13 helicase binding activity ... [https://pubmed.ncbi.nlm.nih.gov/40309067/]
- 49. Monitoring SARS-CoV-2 Nsp13 helicase binding activity using ... [https://pubs.rsc.org/en/content/articlehtml/2025/cb/d4cb00230j]
- 50. High-throughput evaluation of novel WRN inhibitors [https://www.sciencedirect.com/science/article/pii/S2472555225000590]
- 51. Biochemical and Cell Biological Assays to Identify ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5035559/]
- 52. High-throughput evaluation of novel WRN inhibitors [https://pubmed.ncbi.nlm.nih.gov/40865724/]
- 53. What Is the Best Assay for Helicase Enzymes? Formats, ... [https://bellbrooklabs.com/best-assay-helicase-enzymes/?srsltid=AfmBOop5C5AgiyFB4QxlDjAjhL0B7E9Hwx8Nh0pxM7KqBTARSEa-xayA]
- 54. Validation of a High Throughput Screening Assay to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9196137/]
- 55. Structural dynamics of DNA unwinding by a replicative ... [https://www.nature.com/articles/s41586-025-08766-w]
- 56. The Z prime value (Z´) [https://www.bmglabtech.com/en/blog/the-z-prime-value/]
- 57. Empirical Evaluation of a New Method for Calculating ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2394959/]
- 58. - Principles Coefficient of variation [https://influentialpoints.com/Training/coefficient_of_variation-principles-properties-assumptions.htm]
- 59. Z-factor [https://en.wikipedia.org/wiki/Z-factor]
- 60. Use of Coefficient Assessing Variability of Quantitative... of Variation in [https://pmc.ncbi.nlm.nih.gov/articles/PMC130103/]
- 61. Identifying SARS-CoV-2 antiviral compounds by screening ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8286831/]
- 62. What Is the Best Assay for Helicase Enzymes? Formats, ... [https://bellbrooklabs.com/best-assay-helicase-enzymes/?srsltid=AfmBOorKIdtEXGCVIxnMbQQyPsSnCI-cLVvdkpEDpScX6HVuvB_C8W9H]
- 63. Insight into the biochemical mechanism of DNA helicases ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9534331/]
- 64. Development of assays for measuring RNA helicase activity [https://www.news-medical.net/whitepaper/20240514/Advancing-cancer-therapeutics-Development-of-selective-RNA-helicase-inhibitors.aspx]
- 65. Kinetic characterization of three human DExD/H-box RNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11839018/]
- 66. origins of compound-dependent assay interference [https://www.sciencedirect.com/science/article/abs/pii/S1367593110000463]
- 67. Avoiding Fluorescence Assay Interference—The Case for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4840916/]
- 68. Common Challenges in Biochemical Assays and How to ... [https://bellbrooklabs.com/common-challenges-in-biochemical-assays-and-how-to-overcome-them/?srsltid=AfmBOor7IajflpY8fZ_WQ10x5fFGUi1UdMI-lWpLuRQZZe1V8TO1_6MI]
- 69. Enzymes Storage Best Practices to Prevent Pre [https://k2sci.com/blogs/news/enzymes-storage-best-practices-to-prevent-premature-degradation?srsltid=AfmBOopMDG5RnQL0_XOUux-_AilFCLctt9TNMyvJGP1og-L7WK-SmjSt]
- 70. What Is the Best Assay for Helicase Enzymes? Formats, ... [https://bellbrooklabs.com/best-assay-helicase-enzymes/?srsltid=AfmBOookO9GViYxT_Xb4bIlx1RlAt_FC9YCtiKhbSlxbJRbKrYEIp9qM]
- 71. Enzyme Stability - an overview [https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/enzyme-stability]
- 72. Stability Improvement of a Liquid Enzyme Product - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2799593/]
- 73. Reagent Stability and Thawing Guidelines for MiSeq ... [https://knowledge.illumina.com/instrumentation/miseq/instrumentation-miseq-reference_material-list/000001674]
- 74. Real-time fluorescence assays to monitor duplex ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5524973/]
- 75. Hijacking a real time detection thermocycler for enzymology [https://www.sciencedirect.com/science/article/pii/S0300908425000379]
- 76. Enhancing therapeutic antibody profiling: orthogonal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12510943/]
- 77. Orthogonal approach and critical quality attributes for gene ... [https://rrpharmacology.ru/index.php/journal/article/view/738/629]
- 78. Conversion of natural cytokine receptors into orthogonal ... [https://www.nature.com/articles/s41589-025-01986-1]
- 79. Validating a new quantitative assay [https://www.pathologyoutlines.com/topic/managementlabvalidatingnewassay.html]
- 80. New Insights Into DNA Helicases as Druggable Targets for ... [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2018.00059/full]
- 81. Pilot-Scale Screening of Clinically Approved Drugs to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11456206/]
- 82. Development of a fluorescence-based assay for RecBCD ... [https://pubs.rsc.org/en/content/articlehtml/2025/cb/d4cb00291a]
- 83. Using Universal Inhibitor Screening Assays to Accelerate ... [https://bellbrooklabs.com/using-universal-inhibitor-screening-assays/?srsltid=AfmBOoqDC9VGXkYTOmQ1Oe1VAX3ODwkwPtr_4jkvMCxucS_nMTxR3_cV]
- 84. Cellular Thermal Shift Assay ( CETSA ) [https://www.news-medical.net/life-sciences/Cellular-Thermal-Shift-Assay-(CETSA).aspx]
- 85. Determination of Ligand-Target Interaction in vitro by Cellular Thermal... [https://bio-protocol.org/en/bpdetail?id=5047&type=0]
- 86. A superior loading control for the cellular thermal shift assay | Scientific... [https://www.nature.com/articles/s41598-022-10653-7?error=cookies_not_supported&code=4b0604fd-793c-47c0-9fd6-da36c0a76244]
- 87. A fluorescence-based helicase assay - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC4477640/]
- 88. Heli-SMACC: Helicase-targeting SMAll Molecule ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11257486/]
- 89. Heli-SMACC: Helicase-targeting SMAll Molecule Compound ... [https://pubmed.ncbi.nlm.nih.gov/39026851/]
- 90. Identification of MCM2-Interacting Proteins Associated with ... [https://www.mdpi.com/1422-0067/26/3/1020]
- 91. Synthetic Lethality‐Based Targets and Their Exploration in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12381573/]
- 92. A requirement for STAG2 in replication fork progression ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6459917/]
- 93. Cyclin-Dependent Kinase Synthetic Lethality Partners in ... [https://www.mdpi.com/1422-0067/23/7/3555]
- 94. ESMO 2025 – synthetic lethality leaves much to prove [https://www.oncologypipeline.com/apexonco/esmo-2025-synthetic-lethality-leaves-much-prove]