Biochemical Kinase Assays: A Comprehensive Guide to Activity Detection and Phosphorylation Analysis for Drug Discovery
This article provides researchers, scientists, and drug development professionals with a comprehensive guide to biochemical kinase assays, covering foundational principles to advanced applications.
Biochemical Kinase Assays: A Comprehensive Guide to Activity Detection and Phosphorylation Analysis for Drug Discovery
Abstract
This article provides researchers, scientists, and drug development professionals with a comprehensive guide to biochemical kinase assays, covering foundational principles to advanced applications. It explores the critical role of kinases in cellular signaling and disease, detailing established and emerging methodologies for activity detection and phosphorylation analysis. The content addresses common troubleshooting scenarios, optimization strategies for robust assay performance, and frameworks for method validation and comparative analysis to support high-quality data generation in basic research and drug discovery pipelines.
Kinase Fundamentals: Understanding Phosphotransfer and Cellular Signaling Mechanisms
The Central Role of Kinases in Cellular Regulation and Disease Pathogenesis
Protein kinases represent a vast family of enzymes that catalyze the transfer of a phosphate group from adenosine triphosphate (ATP) to specific substrate proteins, primarily on serine, threonine, or tyrosine residues. This process, known as phosphorylation, serves as a fundamental regulatory mechanism controlling virtually every aspect of cellular function, including signal transduction, protein-protein interactions, subcellular localization, and apoptosis [1] [2]. Kinases constitute approximately 2% of the human genome, underscoring their pervasive role in eukaryotic biology [2] [3].
The reversible nature of phosphorylation allows for dynamic cellular responses to external stimuli and internal cues. Dysregulation of kinase activity and phosphorylation dynamics is implicated in numerous disease pathologies, including cancer, diabetes, cardiovascular diseases, and central nervous system disorders [1] [2]. Since the approval of the first kinase inhibitor, imatinib, kinase-targeted therapies have transformed cancer treatment and continue to expand into other therapeutic areas [1] [3].
Biochemical Assays for Kinase Activity Detection
Classification of Kinase Activity Assays
Modern kinase drug discovery relies on biochemical assays that balance sensitivity, throughput, and safety. These assays are generally classified into two main categories: activity assays that directly measure catalytic function, and binding assays that assess inhibitor affinity [1].
Table 1: Major Biochemical Assay Formats for Kinase Activity Detection
| Assay Format | Detection Principle | Key Applications | Throughput | Sensitivity |
|---|---|---|---|---|
| Luminescence-based | Measures ATP consumption or ADP formation (e.g., ADP-Glo, Kinase-Glo) | High-throughput inhibitor screening | High | Moderate-High |
| Fluorescence-based | Uses fluorescent ligands, TR-FRET, or fluorescence polarization | Real-time kinetic measurements, binding studies | High | High |
| Mobility Shift | Separates phosphorylated from non-phosphorylated substrates via capillary electrophoresis | Direct quantitative readouts of kinase activity | Moderate | High |
| Radioactive | Uses radiolabeled [γ-³²P]ATP to measure phosphate transfer | Traditional gold standard, substrate identification | Low | Very High |
| ELISA-based | Quantifies phosphorylated substrates via specific antibody binding | Targeted phosphorylation analysis | Moderate | High |
| Thermal Shift | Measures thermal stability changes upon ligand binding | Binding affinity studies, inhibitor screening | Moderate | Moderate |
Advanced Detection Technologies
Recent technological innovations have significantly enhanced kinase assay capabilities. Fluorescent ligands have emerged as powerful tools that increase assay sensitivity and specificity while enabling real-time monitoring of enzymatic turnover [1]. Fluorescence resonance energy transfer (FRET)-based biosensors allow dynamic monitoring of kinase activity in living cells with spatiotemporal resolution, providing insights into signaling network dynamics [3].
Mass spectrometry-based approaches, particularly liquid chromatography-tandem mass spectrometry (LC-MS/MS), have revolutionized phosphorylation analysis by enabling identification and quantification of thousands of phosphorylation events simultaneously [2] [4]. A novel proteomic kinase activity sensor technique (ProKAS) utilizes tandem arrays of barcoded peptide sensors to enable multiplexed, spatially resolved monitoring of kinase activities via mass spectrometry [5].
Experimental Protocols for Kinase Activity Assessment
Bead-Based Multiplex Kinase Activity Assay
This protocol describes a non-radioactive, bead-based method for detecting kinase activity in vitro, adaptable for multiplexing with Luminex technology [6].
Materials and Reagents
- Avidin-coated microspheres (LumAvidin beads)
- Purified kinase of interest (e.g., PKA, PKC-μ, Akt)
- Biotinylated peptide substrates specific to target kinases
- Phospho-specific primary antibodies (e.g., rabbit anti-phospho-(Ser/Thr) PKA substrate)
- R-PE (phycoerythrin) conjugated secondary antibody (goat anti-rabbit IgG)
- Kinase reaction buffer: 25 mM Tris-HCl (pH 7.5), 5 mM β-glycerol phosphate, 2 mM dithiothreitol (DTT), 0.1 mM Na₃VO₄, 10 mM MgCl₂
- ATP solution (prepared fresh in kinase reaction buffer)
- Bead coating buffer: PBS with 1% BSA (BCB)
- Wash buffer: PBS with 0.05% Tween-20
Procedure
Bead Preparation
- Incubate avidin-coated microspheres with biotinylated peptide substrates (1-5 μg peptide per 5 × 10⁶ beads) for 30 minutes at room temperature with gentle shaking.
- Wash beads twice with PBS containing 0.05% Tween-20 to remove unbound peptide.
- Resuspend beads in kinase reaction buffer at a concentration of 1,000 beads/μL per peptide substrate.
Kinase Reaction
- In a 96-well plate, combine 5 μL of bead suspension (5,000 beads), 5 μL of kinase preparation, and 5 μL of ATP solution (final ATP concentration 10-100 μM).
- Include negative controls without kinase and without ATP.
- Incubate for 30-60 minutes at 30°C with gentle shaking.
Phosphorylation Detection
- Centrifuge plate and carefully remove supernatant.
- Add 50 μL of primary antibody diluted in BCB (typically 1:500 to 1:2000).
- Incubate for 30 minutes at room temperature, protected from light.
- Wash twice with wash buffer.
- Add 50 μL of PE-conjugated secondary antibody (1:2000 dilution in BCB).
- Incubate for 30 minutes at room temperature, protected from light.
- Wash twice with wash buffer and resuspend in 100 μL BCB.
Analysis
- Analyze beads using a Luminex flow cytometry-based instrument.
- Measure median fluorescence intensity (MFI) for each bead set.
- Calculate kinase activity as MFI normalized to negative controls.
Optimization Notes
- Kinase concentration should be titrated to ensure linear reaction kinetics.
- ATP concentration should reflect the Km(ATP) of the specific kinase.
- Antibody concentrations should be optimized for maximum signal-to-noise ratio.
- Multiplexing requires verification that peptide substrates are specific to their intended kinases.
Proteomic Kinase Activity Sensor (ProKAS) Technique
The ProKAS methodology enables multiplexed, spatially resolved monitoring of kinase activity in living cells using mass spectrometry [5].
Materials and Reagents
- ProKAS plasmid constructs with targeting elements (NLS, NES, or organelle-specific signals)
- Cell culture reagents and transfection reagents
- Lysis buffer: 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1% NP-40, with protease and phosphatase inhibitors
- Anti-ALFA affinity beads or alternative affinity resin
- Trypsin for proteolytic digestion
- LC-MS/MS system with appropriate buffers
Procedure
Sensor Design and Construction
- Select 10-15 amino acid peptide sequences representing preferred substrate motifs for kinases of interest.
- Incorporate three or more small amino acid residues at peptide edges for barcoding.
- Flank each kinase substrate motif with arginine residues to ensure proper tryptic cleavage.
- Clone tandem array of sensor peptides into ProKAS vector containing N-terminal eGFP, affinity tag, and targeting element.
Cell Transfection and Treatment
- Transfect cells with ProKAS constructs using standard methods.
- Apply experimental treatments (e.g., kinase inhibitors, activators, genotoxic agents).
- For spatial studies, transfer cells to fractionation or imaging protocols.
Sample Preparation and Affinity Purification
- Lyse cells in appropriate lysis buffer.
- Incubate lysates with anti-ALFA affinity beads for 1-2 hours at 4°C.
- Wash beads thoroughly to remove non-specific interactions.
- Elute bound ProKAS sensors or proceed to on-bead digestion.
Tryptic Digestion and MS Analysis
- Digest samples with trypsin (1:50 enzyme-to-substrate ratio) overnight at 37°C.
- Analyze resulting peptides via LC-MS/MS.
- Use parallel reaction monitoring (PRM) for targeted quantification.
- Identify and quantify both phosphorylated and unphosphorylated sensor peptides.
Data Analysis
- Calculate phosphorylation rate as abundance of phosphorylated sensor peptide normalized to unmodified version.
- Compare phosphorylation across different subcellular compartments based on targeting elements.
- Generate kinetic profiles of kinase activity from time-course experiments.
Validation Steps
- Verify MS detection of phosphorylated peptide sensors.
- Confirm stimulus-dependent regulation of probe phosphorylation.
- Demonstrate specificity of probe phosphorylation for intended kinase using inhibitors or genetic approaches.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Research Reagent Solutions for Kinase Studies
| Reagent Category | Specific Examples | Function and Application |
|---|---|---|
| Fluorescent Ligands | Fluorescent kinase inhibitors, FITC- or Cy-labeled ATP analogs | Enhanced sensitivity for binding assays, real-time monitoring of kinase activity [1] |
| Activity Assay Kits | ADP-Glo, Kinase-Glo | Luminescence-based detection of kinase activity through ATP depletion or ADP formation [1] |
| Phospho-Specific Antibodies | Anti-pSer, Anti-pThr, Anti-pTyr antibodies | Detection of phosphorylated proteins in Western blot, ELISA, and immunofluorescence [7] |
| Specialized Substrates | Phos-tag reagents, Peptide arrays | Phosphorylation site detection without antibodies; profiling kinase specificity [7] [2] |
| Engineered Kinase Systems | Analog-specific (AS) kinase mutants | Specific labeling of direct kinase substrates using bulky ATP analogs [3] |
| Cellular Sensors | FRET-based biosensors, ProKAS constructs | Live-cell monitoring of kinase activity with spatial and temporal resolution [3] [5] |
| Bead-Based Platforms | LumAvidin beads with peptide substrates | Multiplexed kinase activity assessment using flow cytometry [6] |
Structural and Functional Impact of Phosphorylation
Understanding the structural consequences of phosphorylation is essential for elucidating its regulatory functions. A recent global comparative analysis of phosphorylated and non-phosphorylated protein structures revealed that phosphorylation typically induces small, stabilizing conformational changes [8]. The median backbone root mean squared deviation (RMSD) between phosphorylated and non-phosphorylated structures is 1.14 Å, with only 28% of phosphorylation events associated with changes ≥2 Å [8]. This suggests that phosphorylation often fine-tunes protein function rather than inducing dramatic structural rearrangements.
Phosphorylation sites within structured protein domains appear to function through several mechanistic principles:
- Electrostatic effects: Introduction of negative charge alters local electrostatic potential
- Allosteric regulation: Phosphorylation at distal sites modulates functional regions
- Mechanical coupling: Phosphosites show mechanical coupling with functional sites aligned with the domino model of allosteric signal transduction [8]
Notably, phosphorylation within protein kinase domains themselves is associated with significantly larger conformational changes (median RMSD: 1.51 Å) compared to other protein classes, highlighting the importance of phosphorylation in regulating kinase activation states [8].
Visualization of Kinase Signaling and Experimental Workflows
DNA Damage Response Kinase Signaling Pathway
ProKAS Experimental Workflow for Multiplexed Kinase Activity Monitoring
Kinases play a central role in cellular regulation, and their dysregulation underpins numerous disease pathologies. Contemporary biochemical assays for kinase activity detection have evolved from simple radioactive measurements to sophisticated multiplexed platforms that provide unprecedented insights into kinase function, specificity, and cellular localization. The integration of fluorescent technologies, mass spectrometry-based proteomics, and structural biology approaches continues to advance our understanding of phosphorylation networks.
The experimental protocols and reagents described herein provide researchers with robust methodologies for investigating kinase activity in both in vitro and cellular contexts. As kinase-targeted therapies expand beyond oncology into cardiovascular, metabolic, and neurological disorders, these tools will prove increasingly valuable for drug discovery and development. Future directions in kinase research will likely focus on achieving greater spatial and temporal resolution of kinase activity measurements, mapping complete kinase-substrate relationships, and developing more selective therapeutic agents that exploit unique aspects of kinase regulation.
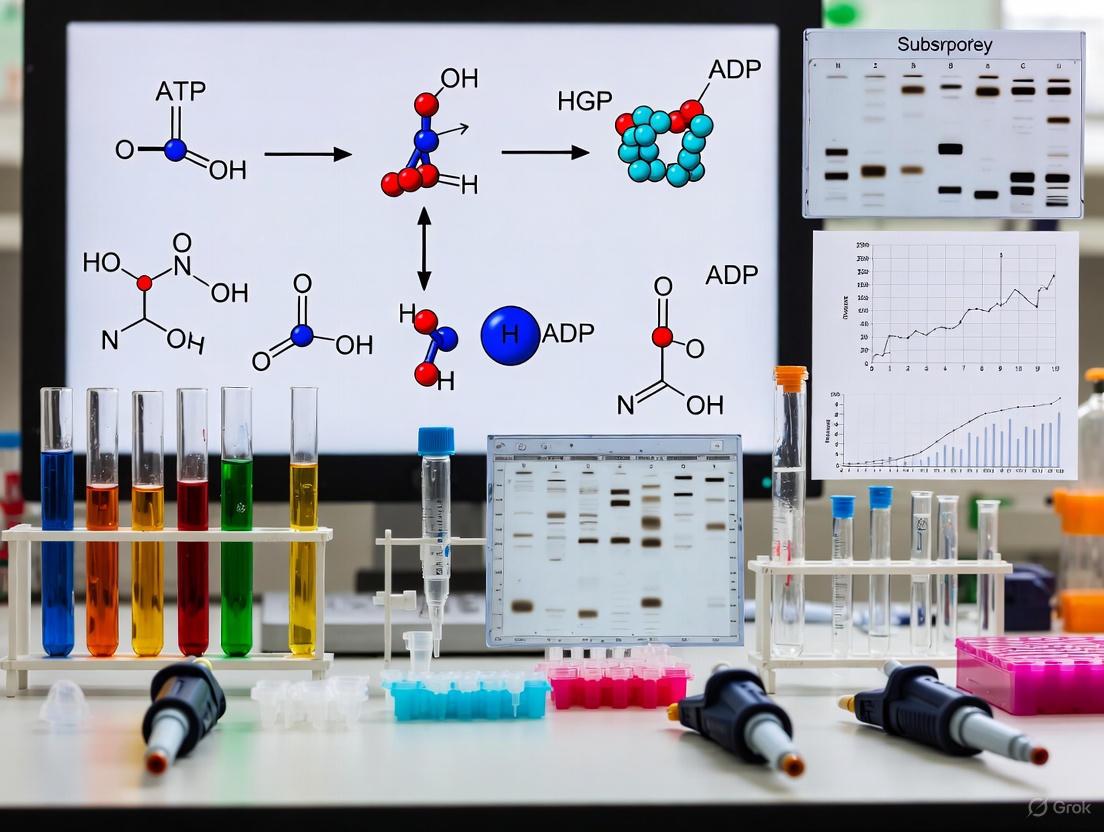
Protein kinases are fundamental enzymes that catalyze the transfer of a gamma-phosphate group from adenosine triphosphate (ATP) to specific serine, threonine, or tyrosine residues on protein substrates. This phosphotransfer reaction represents a crucial regulatory mechanism in cellular signaling, controlling diverse processes including cell cycle progression, growth, differentiation, and metabolism [1] [9]. The catalytic activity of kinases is precisely regulated, and their dysregulation is implicated in numerous diseases, particularly cancer, making them prime targets for therapeutic intervention [1] [10]. Understanding the molecular mechanics of the phosphotransfer reaction—specifically the roles of ATP, magnesium ions, and substrate recognition—is essential for developing robust biochemical assays in drug discovery and basic research.
This application note examines the critical components of kinase-catalyzed phosphotransfer, with emphasis on the indispensable role of magnesium cofactors in facilitating phosphoryl transfer and maintaining structural organization within the active site. We present detailed protocols for assessing kinase activity through multiple biochemical approaches, discuss methodological considerations for accurate measurement, and provide troubleshooting guidance to overcome common experimental challenges. The information presented herein supports ongoing research in kinase biology and inhibitor development by establishing standardized methodologies for interrogating kinase function.
The Essential Role of Magnesium in Phosphotransfer
Magnesium as a Catalytic Cofactor
Magnesium ions are indispensable for kinase catalysis, with an estimated 90% of cellular ATP existing as a complex with Mg²⁺ [11]. The primary function of Mg²⁺ is electrostatic stabilization of the negatively charged phosphate groups of ATP through Lewis acid-base interactions. The high charge density of the Mg²⁺ ion (due to its small ionic radius) attracts electrons from the phosphorus atom, increasing its electrophilicity and making it more susceptible to nucleophilic attack by the substrate hydroxyl group [11]. This process, termed electrostatic catalysis, lowers the activation energy required for phosphoryl transfer.
Most protein kinases require two magnesium ions for optimal catalysis [12]. Kinetic analyses of Cyclin-dependent kinase 2 (CDK2) have demonstrated that simultaneous binding of two Mg²⁺ ions is essential for catalysis of phosphoryl transfer [12]. The first metal ion (MgII) primarily coordinates the α- and β-phosphates of ATP and anchors them to conserved aspartate (Asp145 in CDK2) and asparagine (Asn132) residues within the catalytic cleft. The second ion (MgI) coordinates the β- and γ-phosphates and is crucial for stabilizing the transition state during phosphoryl transfer [12].
Structural Reorganization Induced by Magnesium
Recent structural studies reveal that Mg²⁺ induces significant conformational rearrangements beyond mere substrate neutralization. Research on adenylate kinase demonstrates that Mg²⁺ binding triggers a 30° adjustment in the angle between the phosphate donor and acceptor groups, optimizing geometry for nucleophilic attack [11]. This "reaction angle" of approximately 168° positions the attacking oxygen on the substrate (AMP in adenylate kinase) in line with the target phosphorus atom of the ATP γ-phosphate, facilitating efficient inline displacement [11].
In the reactant state, Mg²⁺ adopts a "capping" coordination with the β- and γ-phosphates of ATP. Following phosphoryl transfer, this transitions to a "bridging" interaction between the β-phosphates of two ADP molecules in the product state [11]. Molecular dynamics simulations indicate that Mg²⁺ binding restricts phosphate group mobility and promotes closure of the glycine-rich loop, excluding water from the active site and creating a favorable environment for catalysis [12]. This structural reorganization demonstrates allosteric regulation beyond direct substrate coordination.
Table 1: Quantitative Effects of Magnesium on Kinase Catalysis
| Parameter | Effect of Magnesium | Experimental System | Reference |
|---|---|---|---|
| Reaction Angle | Optimizes to ~168° for nucleophilic attack | Adenylate kinase crystal structures | [11] |
| Catalytic Rate (k~cat~) | Increases from 0 to 15 s⁻¹ with Mg²⁺ addition (0-7 mM) | pCDK2/Cyclin A | [12] |
| Glycine-rich Loop | Stabilizes closed conformation, excluding water | Molecular dynamics simulations | [12] |
| Product Release | Becomes rate-limiting due to enhanced ADP affinity | CDK2 kinetic analysis | [12] |
| Coordination Geometry | Tetragonal bipyramidal/octahedral with 2.0 Å distance to oxygen ligands | Structural analysis | [11] |
The Magnesium Dilemma in Catalytic Efficiency
While essential for catalysis, the two-metal mechanism presents a kinetic challenge termed the "Mg²⁺ dilemma." Although simultaneous binding of two Mg²⁺ ions dramatically accelerates the chemical step of phosphoryl transfer, this arrangement also cooperatively enhances ADP product affinity, making product release rate-limiting for fully activated CDK2 [12]. Structural analyses reveal that the rigid ADP·2Mg²⁺ product complex closely resembles the transition state geometry, rationalizing the strong product inhibition observed in many kinase systems [12]. This dual functionality means the same Mg²⁺ binding site functions as both an activator (during phosphoryl transfer from ATP) and an inhibitor (during product release), requiring careful evolutionary tuning of metal affinity to balance catalytic efficiency with timely product dissociation.
Experimental Approaches for Kinase Activity Assessment
Biochemical Assay Formats
Multiple biochemical assay formats have been developed to quantify kinase activity, each with distinct advantages and limitations. These can be broadly categorized into activity assays that directly measure catalytic function and binding assays that assess inhibitor interactions [1].
Radioactive assays represent the historical gold standard, utilizing γ-³²P or ³³P-ATP to directly label substrate proteins or peptides. The phosphorylated products are typically captured on filter membranes (HotSpot assay) or detected using scintillation counting (33PanQinase assay) [13]. These formats provide direct measurement of enzyme activity with minimal false positives/negatives and are adaptable to diverse substrates. However, they require special handling procedures for radioactivity disposal and safety [13].
Luminescence-based assays (e.g., ADP-Glo) quantify ADP formation as a surrogate of kinase activity by coupling ATP depletion to luciferase-generated luminescence signals [1]. These homogeneous assays are suitable for high-throughput screening and eliminate radioactivity concerns while providing good sensitivity across miniaturized formats.
Fluorescence-based assays employ various mechanisms including:
- TR-FRET/HTRF: Measure energy transfer between antibody-bound fluorophores recognizing phosphorylated substrates
- Fluorescence polarization: Detect changes in rotational mobility upon phosphorylation
- Mobility shift assays: Separate phosphorylated from non-phosphorylated substrates via capillary electrophoresis [1]
These non-radioactive formats generally offer higher throughput and better safety profiles, though they may be susceptible to compound interference through fluorescence quenching or autofluorescence [1].
Table 2: Comparison of Kinase Activity Assay Formats
| Assay Format | Detection Method | Throughput | Sensitivity | Advantages | Limitations |
|---|---|---|---|---|---|
| Radioactive | ³³P-ATP incorporation | Medium | High | Direct activity measurement; Low false positives | Radioactive handling; Special disposal |
| ADP-Glo | Luminescence from ADP detection | High | Medium | Non-radioactive; Homogeneous format | Indirect measurement; Coupled enzyme system |
| TR-FRET | FRET with time-resolution | High | Medium-High | Homogeneous; Low background | Antibody-dependent; Signal interference |
| Mobility Shift | Capillary electrophoresis | Medium | High | Direct phosphorylation measurement | Instrument-intensive; Lower throughput |
| ELISA | Colorimetric/chemiluminescent | Medium | High | Quantitative; Specific | Plate-based; Multiple washing steps |
Phosphorylation Detection Methods
Direct detection of phosphorylated proteins provides complementary information to activity assays, enabling assessment of specific phosphorylation events in complex biological systems.
Western Blotting using phospho-specific antibodies remains the most widely used method for detecting protein phosphorylation [9]. This technique separates proteins by molecular weight before transfer to membranes and immunodetection with phosphorylation-state-specific antibodies. A significant advantage is the ability to normalize phospho-protein levels to total protein expression using cognate antibodies, controlling for loading variations [9]. Automated capillary Western systems (e.g., Simple Western) enhance reproducibility, sensitivity, and throughput while requiring minimal sample volumes (3 μL) [9].
Enzyme-Linked Immunosorbent Assay (ELISA) provides quantitative measurement of specific phosphorylation events through sandwich immunoassays, typically employing a capture antibody against the target protein and a phospho-specific detection antibody [9]. Phospho-ELISAs offer superior quantification compared to Western blotting, higher sensitivity for detecting low-abundance proteins, and compatibility with high-throughput microplate formats [9].
Intracellular Flow Cytometry enables single-cell analysis of phosphorylation events within heterogeneous cell populations, allowing researchers to correlate signaling states with surface marker expression without physical separation [9]. This approach is particularly valuable for identifying phosphorylation responses in rare cell subsets and for monitoring signaling heterogeneity within populations.
Phosphoproteomic Approaches utilize mass spectrometry to globally identify and quantify phosphorylation sites, providing systems-level views of kinase signaling networks [10]. Computational methods like KSEA (Kinase-Substrate Enrichment Analysis) and PTM-SEA then infer kinase activities from the phosphorylation patterns of their known substrates [10]. While powerful for discovery, these approaches require specialized instrumentation and computational expertise.
Detailed Experimental Protocols
Protocol 1: Immunoblotting for Endogenous Kinase Activity (Plant SnRK2)
This protocol adapts methodology for detecting SnRK2 kinase activity in plants [14] for general application to endogenous kinase assessment.
Materials and Reagents
- Lysis buffer (e.g., RIPA buffer with fresh protease and phosphatase inhibitors)
- Protein extraction: Prechilled mortar and pestle or tissue homogenizer
- Phospho-specific antibody against target kinase activation loop
- Total target kinase antibody for normalization
- HRP-conjugated secondary antibodies
- Enhanced chemiluminescence (ECL) substrate
- PVDF or nitrocellulose membrane
- Electrophoresis and transfer apparatus
Procedure
Sample Preparation:
- Homogenize tissue or cells in ice-cold lysis buffer (100 mg tissue/mL buffer)
- Centrifuge at 14,000 × g for 15 minutes at 4°C
- Transfer supernatant to fresh tubes and quantify protein concentration
SDS-PAGE and Transfer:
- Separate 20-50 μg total protein by SDS-PAGE (8-12% gel)
- Transfer to PVDF membrane using wet or semi-dry transfer systems
Immunoblotting:
- Block membrane with 5% non-fat dry milk in TBST for 1 hour
- Incubate with primary phospho-specific antibody (dilution per manufacturer's recommendation) in blocking buffer overnight at 4°C
- Wash membrane 3 × 10 minutes with TBST
- Incubate with HRP-conjugated secondary antibody (1:2000-1:5000) for 1 hour at room temperature
- Wash 3 × 10 minutes with TBST
- Develop with ECL substrate and image
Membrane Reprobing:
- Strip membrane with mild stripping buffer
- Re-block and probe with total target kinase antibody
- Normalize phospho-signal to total kinase signal
Data Interpretation
- Calculate ratio of phospho-signal to total kinase signal for each sample
- Compare relative kinase activation across experimental conditions
- Include positive and negative controls for kinase activation status
Protocol 2: Radioactive In-Gel Kinase Assay
This protocol based on SnRK2 assessment [14] provides direct measurement of kinase activity following native electrophoresis.
Materials and Reagents
- Kinase reaction buffer (20 mM HEPES pH 7.4, 10 mM MgCl₂, 1 mM DTT, 1 mM CaCl₂)
- Myelin basic protein (0.5 mg/mL) or other substrate copolymerized in the gel
- [γ-³²P]ATP or [γ-³³P]ATP (10 μCi per sample)
- Cold ATP (100 μM final concentration)
- Precast native polyacrylamide gels or components for casting Laemmli gels without SDS
- Fixing solution (10% acetic acid, 25% isopropanol)
- Phosphorimager screen and scanner or X-ray film
Procedure
Native Gel Electrophoresis:
- Prepare samples in non-denaturing, non-reducing loading buffer
- Separate proteins by electrophoresis through native PAGE gel containing substrate protein at 4°C
- Do not boil samples or include SDS or reducing agents
Kinase Reaction In-Gel:
- Wash gel 2 × 30 minutes with renaturation buffer (25 mM Tris pH 8.0, 1 mM DTT, 0.1% Triton X-100) to remove urea and renature proteins
- Equilibrate gel in kinase reaction buffer for 30 minutes
- Incubate gel in kinase reaction buffer containing 1 μCi/mL [γ-³²P]ATP and 100 μM cold ATP for 1-2 hours at room temperature with gentle agitation
- Include control gels without substrate protein to distinguish specific phosphorylation
Detection:
- Stop reaction by transferring gel to fixing solution for 1-2 hours
- Wash extensively with fixing solution to remove unincorporated radioactivity
- Dry gel and expose to phosphorimager screen or X-ray film
- Quantify band intensity corresponding to kinase activity
Data Interpretation
- Band intensity correlates with kinase activity
- Compare migration position with protein standards for molecular weight estimation
- Normalize to total protein loading controls when possible
Protocol 3: Band-Shift Assay for Tagged Kinases
This protocol assesses activation-induced mobility shifts of epitope-tagged kinases due to autophosphorylation [14].
Materials and Reagents
- Transgenic lines expressing tagged kinase of interest
- Phosphate-affinity binding reagents (e.g., Phos-tag)
- Modified Laemmli sample buffer
- Special SDS-PAGE gels with phosphate-binding compounds
- Transfer and immunoblotting equipment
- Antibody against epitope tag
Procedure
Sample Preparation:
- Extract proteins from transgenic tissue expressing tagged kinase
- Prepare samples in Laemmli buffer without EDTA (can interfere with phosphate binding)
Phos-tag Gel Electrophoresis:
- Prepare or purchase SDS-PAGE gels containing 25-100 μM Phos-tag reagent and appropriate divalent cations
- Conduct electrophoresis at low voltage (≤100V) with cooling to maintain separation resolution
- Include control samples with phosphatase treatment to confirm phosphorylation-dependent shifts
Detection:
- Transfer proteins to membrane using standard Western transfer protocols
- Probe with antibody against the epitope tag
- Develop using standard ECL detection
Data Interpretation
- Multiple bands indicate different phosphorylation states
- Upward shifts correspond to increased phosphorylation
- Compare band patterns across experimental conditions to assess kinase activation status
Research Reagent Solutions
Table 3: Essential Research Reagents for Kinase Studies
| Reagent Category | Specific Examples | Function/Application | Considerations |
|---|---|---|---|
| Magnesium Salts | MgCl₂, MgSO₄ | Essential kinase cofactor; Optimize concentration (1-20 mM) | Concentration affects kinetics and rate-limiting steps |
| ATP Solutions | ATP disodium salt | Phosphate donor substrate; Typical range 1-100 μM for assays | Physiological concentration ~1 mM; Use appropriate Km |
| Protease Inhibitors | PMSF, protease inhibitor cocktails | Prevent sample degradation during extraction | Include in all lysis buffers |
| Phosphatase Inhibitors | Sodium fluoride, β-glycerophosphate, sodium orthovanadate | Preserve phosphorylation status | Essential for maintaining in vivo phosphorylation states |
| Phospho-specific Antibodies | Anti-phospho-S175-SnRK2 [14] | Detect activation loop phosphorylation | Validate specificity with phosphorylation-deficient mutants |
| Kinase Substrates | Myelin basic protein, histone H1 | Generic substrates for activity assays | Specific peptide substrates also available |
| Detection Reagents | ECL substrate, fluorescent secondaries | Signal generation for immunodetection | Match detection method to instrument capabilities |
| Radioactive Labels | [γ-³²P]ATP, [γ-³³P]ATP | Direct detection of phosphorylated products | Requires radiation safety protocols |
| Tagging Systems | HA, FLAG, GFP tags | Epitope tags for kinase detection and purification | Minimal tags reduce perturbation of native function |
Troubleshooting and Optimization
Common Experimental Challenges
Low Signal Intensity in kinase assays may result from insufficient kinase activity, suboptimal Mg²⁺ concentrations, improper ATP levels, or ineffective antibodies. To address this, titrate Mg²⁺ concentrations (1-20 mM) while maintaining physiological relevance (~1 mM ATP) [12] [11]. Validate phospho-antibodies with positive controls including constitutively active kinases or λ-phosphatase-treated negative controls [9]. Ensure fresh phosphatase inhibitors are included in extraction buffers to preserve phosphorylation states.
High Background Signal often stems from non-specific antibody binding, incomplete blocking, or insufficient washing. Optimize blocking conditions using 3-5% BSA or non-fat dry milk with 0.1% Tween-20. Include no-primary antibody controls and pre-absorb secondary antibodies if needed. For radioactive assays, increase washing stringency and include substrate-only controls.
Variable Results between replicates can arise from inconsistent cell lysis, protein degradation, or assay conditions. Standardize culture conditions, lysis protocols, and protein quantification methods. Aliquot and store reagents to minimize freeze-thaw cycles. Include internal controls across experiments to normalize for inter-assay variability.
Magnesium and Buffer Optimization
The divalent cation concentration represents a critical optimization parameter for kinase assays. While Mg²⁺ is essential for catalysis, excess Mg²⁺ can inhibit certain kinases through stabilization of the ADP product complex [12]. Titrate Mg²⁺ concentrations from 0.5-20 mM while monitoring both initial velocity and endpoint measurements. Consider that some kinases may utilize Mn²⁺ as an alternative cofactor, though coordination geometry and efficacy may differ [12].
Maintain physiological pH (typically 7.0-7.5) using appropriate buffers (HEPES, Tris) with consideration of temperature-sensitive pKa values. Include reducing agents (DTT, β-mercaptoethanol) to prevent oxidation of cysteine residues, but note that high concentrations may interfere with certain detection methods. Optimize DMSO concentrations (typically <1%) when testing hydrophobic inhibitors to minimize solvent effects on enzyme activity [1].
The phosphotransfer reaction represents a complex biochemical process requiring precise coordination between ATP, magnesium cofactors, and protein substrates. Understanding the dual role of magnesium in both facilitating phosphoryl transfer and potentially limiting catalytic efficiency through product inhibition is essential for designing physiologically relevant kinase assays [12] [11]. The experimental approaches detailed herein provide researchers with multiple pathways for assessing kinase activity, from traditional radioactive methods to modern non-radioactive formats, each with distinct advantages for specific applications.
As kinase drug discovery advances, the integration of robust biochemical assays with structural insights into metal coordination and substrate recognition will continue to drive innovation in therapeutic development. The protocols and troubleshooting guidance presented in this application note establish a foundation for standardized kinase assessment, enabling more accurate characterization of kinase function and inhibition across diverse research contexts.
Kinase Activity Assessment Workflow
Magnesium's Dual Role in Kinase Catalysis
Protein kinases represent one of the largest and most functionally diverse enzyme families in the human genome, playing indispensable roles in regulating cellular processes through phosphorylation [15]. These enzymes catalyze the transfer of a phosphate group from ATP to specific amino acid residues on target proteins, thereby modulating protein function, localization, and stability [10]. The human genome encodes approximately 540 kinases that regulate an estimated 350,000 phosphorylation sites across 20,000 proteins, forming complex signaling networks that control fundamental biological processes including proliferation, differentiation, metabolism, and apoptosis [10] [15].
Kinases are categorized based on their substrate specificity, with three major classes dominating the phospho-regulatory landscape: serine/threonine kinases (STKs), tyrosine kinases (TKs), and dual-specificity kinases capable of phosphorylating both serine/threonine and tyrosine residues. STKs constitute the most abundant class, accounting for over 70% of the human kinome and targeting serine and threonine residues specifically [15]. Tyrosine kinases, while fewer in number, play critical roles in growth factor signaling and oncogenesis. Dual-specificity kinases represent a specialized group that bridges signaling pathways through their ability to recognize multiple residue types, with prominent examples including the mitogen-activated protein kinase kinases (MAP2Ks) and dual-specificity tyrosine-phosphorylation-regulated kinases (DYRKs) [16] [17].
The clinical relevance of kinases extends across human diseases, including cancer, neurodegenerative disorders, metabolic conditions, and infectious diseases [10] [15]. Kinases represent one of the most targeted protein families for therapeutic intervention, with over seventy small-molecule kinase inhibitors approved by the FDA since 2001 [15]. Beyond human biology, pathogenic bacteria also harbor eukaryotic-like serine/threonine kinases that contribute to virulence and antibiotic tolerance, broadening the therapeutic potential of kinase-targeted strategies to include antimicrobial applications [18] [15].
Table 1: Major Kinase Classes and Their Characteristics
| Kinase Class | Target Residues | Representative Families | Key Functional Roles |
|---|---|---|---|
| Serine/Threonine Kinases | Serine, Threonine | MAPK, CDK, Akt, mTOR [15] | Cell cycle progression, metabolism, stress response [15] |
| Tyrosine Kinases | Tyrosine | EGFR, VEGFR, Src [19] | Growth factor signaling, oncogenesis [19] |
| Dual-Specificity Kinases | Serine, Threonine, Tyrosine | MAP2K/MEK, DYRK, IKK2 [20] [16] [17] | Signaling cascade integration, stress response, developmental regulation [20] [16] [17] |
Experimental Approaches for Kinase Activity Analysis
Phosphoproteomics and Kinase Activity Inference
Mass spectrometry (MS)-based phosphoproteomics has revolutionized the systematic analysis of kinase networks by enabling large-scale identification and quantification of phosphorylation events [10]. Modern MS technologies can measure up to 50,000 unique phosphopeptides spanning over 75% of all cellular proteins, providing comprehensive snapshots of the phosphoproteome that reflect kinase and phosphatase activities [10]. To infer kinase activities from phosphoproteomics data, numerous computational methods have been developed that utilize kinase-substrate libraries to connect observed phosphorylation patterns to upstream kinase regulators.
The benchmarKIN framework provides a standardized evaluation platform for kinase activity inference methods, incorporating both perturbation-based and tumor-based benchmarking approaches [10]. Perturbation-based evaluation assesses how accurately methods identify known perturbed kinases (e.g., through inhibitor treatments or genetic manipulations), while tumor-based evaluation utilizes multi-omics datasets from clinical samples to identify highly active or inactive kinases in disease contexts [10]. Key computational methods for kinase activity inference include:
- PTM-SEA: Applies single-sample gene set enrichment analysis to phosphorylation data [10]
- KSEA: Calculates z-scores based on aggregation of phosphorylation site levels for known kinase targets [10]
- KARP: Ranks kinase activities using phosphoproteomics data through specialized algorithms [10]
- VIPER: Uses Virtual Inference of Protein-activity by Enriched Regulon analysis to deduce kinase activities [10]
Performance evaluations reveal that while most computational methods perform similarly, the choice of kinase-substrate library significantly impacts inference accuracy, with combinations of manually curated libraries demonstrating superior performance [10]. Adding predicted kinase-substrate interactions from tools like NetworKIN can enhance coverage and improve inference capabilities, particularly for less-studied kinases [10].
Biosensor-Based Binding Analysis
Biosensor technologies have emerged as powerful tools for characterizing kinase-inhibitor interactions, providing critical thermodynamic and kinetic parameters that inform drug discovery efforts [21]. Optical biosensing platforms including surface plasmon resonance (SPR), reflectometric interference spectroscopy (RIfS), and biolayer interferometry (BLI) enable real-time monitoring of binding events between kinases and small molecule inhibitors [21]. These approaches provide valuable information on binding affinity, association rates (k~on~), and dissociation rates (k~off~) using minimal sample amounts, making them particularly valuable for early-stage drug screening [21].
Intelligent combinations of biosensor methods with other analytical techniques can provide comprehensive characterization of kinase-inhibitor interactions, helping to elucidate specificity profiles and mechanism of action [21]. The kinetic parameters obtained through these methods are increasingly recognized as critical predictors of in vivo efficacy and safety profiles, as compounds with slow dissociation rates often demonstrate prolonged target engagement and potentially improved therapeutic indices [21].
Structural and Molecular Dynamics Approaches
Structure-based drug discovery utilizing molecular docking and molecular dynamics (MD) simulations has become a central strategy for identifying and optimizing kinase inhibitors [15]. Molecular docking predicts binding poses and affinities of small molecules to kinase structures, facilitating virtual screening of large chemical libraries, while MD simulations capture the time-resolved flexibility of kinases and their complexes with ligands [15].
Recent methodological advances include automated MD workflows, machine learning-driven interaction fingerprinting frameworks, and hybrid docking-MD pipelines that enhance throughput and reproducibility [15]. These approaches are particularly valuable for addressing key challenges in kinase drug discovery, including selectivity against conserved ATP-binding pockets, predicting resistance mutation effects, and characterizing allosteric binding sites [15]. The integration of physics-based simulations with enhanced sampling and machine learning is transforming MD from a descriptive technique into a quantitative component of modern kinase drug discovery pipelines [15].
Diagram 1: Experimental Workflow for Kinase Activity Analysis. This workflow illustrates the integrated computational and experimental approach for kinase activity assessment, highlighting key steps from data acquisition to functional characterization.
Application Notes and Experimental Protocols
Serine/Threonine Kinase Analysis: Bacterial STK Profiling
Background: Bacterial serine/threonine kinases (bSTKs) represent promising targets for antibacterial therapies due to their roles in virulence, cellular development, and antibiotic resistance [18]. Unlike their eukaryotic counterparts, bSTKs have remained relatively understudied, necessitating systematic approaches for their classification and functional characterization.
Protocol: Constraint-Based Classification of bSTK Families
Sequence Identification and Alignment:
- Generate initial alignment profile using experimentally validated bSTKs (e.g., 43 reference sequences)
- Utilize profile-based alignment tools (MAPGAPS) to identify bSTKs from public databases (RefSeq NR, UniProtKB)
- Apply quality filters to ensure kinase domain integrity
Constraint-Based Clustering:
- Implement optimal multiple-category Bayesian Partitioning with Pattern Selection (omcBPPS) algorithm
- Classify aligned sequences into hierarchical categories based on co-conserved patterns
- Build family-specific alignment profiles to refine classification
Family Validation and Characterization:
- Assess conservation of hallmark motifs (VAIK, HRD, DFG) to identify canonical kinases vs. pseudokinases
- Analyze taxonomic distribution across bacterial phyla
- Identify family-specific insertions, deletions, and regulatory domains
Key Results: This approach has successfully classified nearly 300,000 bSTK sequences into 42 distinct families (35 canonical kinase and 7 pseudokinase families) [18]. Actinobacteria exhibited the most diverse STK repertoire with 13 families, while the KAPD family emerged as the most prominent with over 55,000 sequences, including the well-characterized Mycobacterium tuberculosis PknB kinase [18].
Technical Considerations: Approximately 50% of sequences in seven bSTK families show variations in catalytic triad residues essential for phosphotransferase activity, classifying them as pseudokinases with potential regulatory functions [18]. These families display characteristic substitutions: VAIK Lys substitution occurs in all seven pseudokinase families, HRD Asp substitution in six families, and DFG Asp substitution in multiple families [18].
Dual-Specificity Kinase Characterization: IKK2 Mechanism Analysis
Background: IKK2/IKKβ, a prototypical dual-specificity kinase in vertebrates, plays critical roles in NF-κB activation through phosphorylation of IκBα at serines 32 and 36 [20]. Recent evidence indicates that IKK2 also autophosphorylates at tyrosine residues, expanding its functional repertoire beyond traditional serine/threonine kinase activity.
Protocol: Assessing Dual-Specificity Autophosphorylation
Recombinant Protein Preparation:
- Express and purify full-length IKK2 using baculovirus or bacterial expression systems
- Optional: Co-express with regulatory subunit NEMO to assess complex formation effects
In Vitro Kinase Assays:
- Incubate IKK2 (1-2 μg) with ATP (10-100 μM) in kinase buffer (20-30 minutes, 30°C)
- Include controls: kinase-dead mutant (K44M), ATP-competitive inhibitors
- For autophosphorylation detection: Use γ-32P-ATP or phospho-specific antibodies
Site-Specific Mutational Analysis:
- Generate point mutations at key residues (Y169F, S177E/S181E)
- Compare autophosphorylation and substrate phosphorylation capabilities
- Assess phospho-relay activity in presence of ADP
Functional Validation:
- Measure IκBα phosphorylation at S32/S36 using phospho-specific antibodies
- Quantify NF-κB activation in cellular models (e.g., MEF cells reconstituted with IKK2 mutants)
- Monitor downstream transcriptional activity through reporter assays
Key Findings: IKK2 demonstrates dual-specificity through autophosphorylation at Y169, which is essential for subsequent phosphorylation of IκBα at S32 [20]. The conserved ATP-contacting residue K44 is critical for both autophosphorylation and substrate phosphorylation activities [20]. Interestingly, fully autophosphorylated IKK2 can transfer phosphate groups to IκBα in the presence of ADP but absence of ATP, suggesting a unique phospho-relay mechanism distinct from conventional γ-phosphate transfer from ATP [20].
Technical Applications: This protocol enables characterization of atypical kinase mechanisms and identification of allosteric regulatory sites that may be targeted for therapeutic intervention in inflammatory diseases and cancers involving NF-κB pathway dysregulation.
Kinase Inhibitor Screening: Transport and Toxicity Assessment
Background: Tyrosine kinase inhibitors (TKIs) represent a rapidly expanding class of targeted therapeutics, but their unpredictable pharmacokinetic profiles and hepatotoxicity present clinical challenges [19]. Understanding transporter-mediated uptake mechanisms is essential for predicting TKI disposition and toxicity.
Protocol: Competitive Counterflow (CCF) Assay for Transporter Screening
Cell Culture Preparation:
- Maintain HEK293 cells overexpressing OATP1B1 in DMEM + 10% FBS
- Seed cells on poly-D-lysine-coated 96-well plates (2,500 cells/well)
- Induce OATP1B1 expression with 1 μg/mL doxycycline 24 hours pre-assay
CCF Assay Setup:
- Preincubate cells with 0.01 μmol/L 3H-labeled estradiol-17β-glucuronide (EβG) in serum-free medium (1 hour, room temperature)
- Spike wells with test TKIs (0.1-10 mmol/L) or controls (DMSO vehicle, EβG positive control, glucose negative control)
- Incubate 30 minutes to allow competitive displacement
Sample Processing and Analysis:
- Terminate assay with three PBS washes (4°C)
- Solubilize cells with 1% Triton X-100 in PBS (2 hours, room temperature)
- Transfer lysate to scintillation plates, add MicroScint-PS fluid
- Quantify radioactivity using microplate scintillation counter
- Normalize counts to total protein content
Validation Assays:
- Assess VEGFR2 target engagement in OATP1B1-expressing vs. control cells
- Perform molecular docking studies to visualize TKI binding to OATP1B1
- Evaluate in vivo liver-to-plasma ratios in transporter-deficient mouse models
Key Results: Screening of 62 FDA-approved TKIs identified 13 as putative OATP1B1 substrates, including pazopanib [19]. OATP1B1-mediated uptake correlated with hepatotoxicity, as demonstrated by reduced liver-to-plasma ratios and diminished hepatotoxicity in transporter-deficient mice [19].
Technical Applications: The CCF assay provides a robust platform for identifying transporter substrates while overcoming limitations of traditional uptake assays, particularly nonspecific extracellular membrane binding that complicates TKI screening [19]. This approach enables systematic evaluation of hepatic uptake mechanisms underlying TKI disposition and toxicity.
Table 2: Key Experimental Methods for Kinase Analysis
| Method Category | Specific Techniques | Key Applications | Technical Considerations |
|---|---|---|---|
| Phosphoproteomic Analysis | LC-MS/MS, enrichment strategies [10] [22] | Kinase activity inference, substrate identification [10] | Requires kinase-substrate libraries; coverage limitations [10] |
| Biosensor Technologies | SPR, RIfS, BLI [21] | Inhibitor binding kinetics and affinity [21] | Low sample consumption; provides thermodynamic parameters [21] |
| Computational Inference | PTM-SEA, KSEA, KARP, VIPER [10] | Kinase activity profiling from phosphoproteomics [10] | Performance depends on library choice; combination approaches recommended [10] |
| Cellular Transport Assays | Competitive counterflow (CCF) [19] | Transporter substrate identification [19] | Overcomes nonspecific binding issues; suitable for TKIs [19] |
| Molecular Modeling | Docking, MD simulations [15] | Inhibitor design, mechanism elucidation [15] | Addresses conformational flexibility; computational resource-intensive [15] |
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for Kinase Studies
| Reagent/Category | Specific Examples | Function/Application | Technical Notes |
|---|---|---|---|
| Kinase-Substrate Libraries | PhosphoSitePlus, SIGNOR, Phospho.ELM [10] | Kinase activity inference from phosphoproteomics [10] | Manually curated databases superior to predicted alone; combination recommended [10] |
| Predictive Algorithms | NetworKIN [10] | Expanding kinase-substrate coverage [10] | Boosts assessable kinases when combined with curated libraries [10] |
| Dual-Specificity Kinase Inhibitors | GSK-626616 [17] | DYRK family inhibition; cilium studies [17] | Leads to cilium elongation and morphological defects [17] |
| Biosensor Platforms | SPR, RIfS, BLI systems [21] | Kinetic analysis of kinase-inhibitor interactions [21] | Provides binding affinity (K~D~) and kinetics (k~on~, k~off~) [21] |
| Transporter Assay Systems | OATP1B1-overexpressing HEK293 cells [19] | Hepatic uptake assessment for TKIs [19] | CCF format reduces nonspecific binding artifacts [19] |
| Molecular Modeling Suites | Schrödinger Maestro, AutoDock, GROMACS [15] [19] | Structure-based kinase inhibitor design [15] | Docking for pose prediction; MD for dynamics and stability [15] |
| Phosphosite Analysis Tools | PhoSiteformer, CSPred [22] | Identification of critical regulatory phosphosites [22] | Combines sequence and phosphoproteomics data; bimodal fusion [22] |
Diagram 2: Kinase Signaling Network Integration. This diagram illustrates how dual-specificity kinases integrate signals from serine/threonine and tyrosine kinase pathways to regulate diverse cellular processes, highlighting their central position in signaling networks.
The expanding toolkit for kinase analysis, encompassing phosphoproteomic profiling, biosensor technologies, computational inference, and specialized cellular assays, continues to deepen our understanding of kinase diversity and function. The integration of these approaches enables comprehensive characterization of kinase networks across the major classes of serine/threonine, tyrosine, and dual-specificity kinases. As methodological innovations continue to emerge, particularly in computational prediction and structural modeling, researchers are better equipped to address longstanding challenges in kinase drug discovery, including selectivity, resistance, and toxicity prediction. The protocols and application notes detailed herein provide a framework for advancing kinase research in both basic science and therapeutic development contexts.
The Inhibitor of κB Kinase-complex (IKK-complex) serves as a central signaling node in the canonical NF-κB pathway, governing cellular responses to inflammation and pathogenic signals [23] [20]. This complex comprises two catalytic subunits, IKK1 (IKKα) and IKK2 (IKKβ), along with the essential regulatory scaffolding protein NEMO (IKKγ) [23] [24]. Among these, IKK2 performs the critical function of phosphorylating the inhibitor of κBα (IκBα) at two specific serine residues (S32 and S36), which marks IκBα for ubiquitin-dependent proteasomal degradation [23] [20]. This degradation liberates the transcription factor NF-κB, allowing its translocation to the nucleus to activate gene expression programs essential for inflammatory and immune responses [23] [24].
IKK2 is a multidomain protein featuring a kinase domain (KD), a ubiquitin-like domain (ULD), a scaffold dimerization domain (SDD), and a C-terminal NEMO-binding domain (NBD) [23] [20]. Its activity is tightly regulated through phosphorylation of two serine residues (S177 and S181) within its activation loop (AL) [23] [24]. Traditionally classified as a serine/threonine kinase, recent fundamental research has revealed that IKK2 exhibits unexpected dual specificity, capable of autophosphorylating at tyrosine residues in addition to its known serine autophosphorylation activities [23] [20] [24]. This newly discovered mechanism, which involves an unconventional phospho-relay from tyrosine-phosphorylated IKK2 to its substrate IκBα, challenges the established paradigm of phosphate transfer directly from ATP to substrate in eukaryotic protein kinases [20] [24].
Key Findings on IKK2 Dual-Specificity Autophosphorylation
IKK2 as a Dual-Specificity Kinase
The seminal finding from recent studies demonstrates that IKK2 undergoes multisite autophosphorylation not only at its canonical activation loop serines (S177/S181) but also at tyrosine residues, classifying it as a dual-specificity kinase [23] [24]. This tyrosine autophosphorylation is functionally dependent on prior phosphorylation of S177/S181, indicating a hierarchical activation mechanism [23] [20]. Biochemical analyses of catalytically competent IKK2 revealed its capacity for autophosphorylation at tyrosine residues proximal to the active site, in addition to its established serine phosphorylation activities [23]. This finding substantially advances our understanding of IKK2's mechanism of action and could redefine its specificity in propagating cellular inflammatory responses [23] [24].
Critical Role of Y169 in Substrate Phosphorylation
A key autophosphorylation site identified is Y169, located at the DFG+1 position (DLG in IKK1/2) near the active site [23] [20]. Mutation of Y169 to phenylalanine (Y169F) renders IKK2 incapable of phosphorylating S32 within its IκBα substrate [23] [24]. Cellular studies corroborated this finding, showing that signal-responsive phosphorylation of IκBα upon TNF-α treatment was severely diminished in mouse embryonic fibroblast (MEF) cells reconstituted with IKK2-Y169F compared to wild-type IKK2 [23] [20]. This highlights the essential role of Y169 autophosphorylation in IKK2's specific kinase activity toward its primary substrate.
Essential Function of the Conserved K44 Residue
The evolutionarily conserved ATP-contacting residue K44 in IKK2 proved critical for maintaining phosphorylation specificity [23] [20]. Mutation of K44 to methionine (K44M) resulted in complete loss of autophosphorylation at both S177/S181 and tyrosine residues, with consequent inability to phosphorylate IκBα at S32/S36 [23] [20]. Interestingly, IKK2-K44M retained non-specific kinase activity, phosphorylating IκBα at other residues outside the signal-responsive S32/S36 sites, particularly within the C-terminal PEST region [23] [20]. This demonstrates that K44 is indispensable for signal-responsive substrate specificity but not for general catalytic activity.
Unconventional Phospho-Relay Mechanism
Perhaps the most striking finding is the evidence for a phospho-relay mechanism from autophosphorylated IKK2 to IκBα [20] [24]. Researchers observed that fully autophosphorylated IKK2 could transfer phosphate groups to IκBα in the absence of exogenous ATP when ADP was present in the reaction [20] [24]. This suggests the existence of a unique, likely transient, autophosphorylated form of IKK2 that serves as a phosphate donor specifically for S32/S36 of IκBα [20]. This mechanism contrasts fundamentally with the conventional transfer of γ-phosphate groups directly from ATP to substrate observed in eukaryotic protein kinases [20].
Table 1: Key Functional Sites in IKK2 and Effects of Mutations
| Site | Position | Mutation | Autophosphorylation | IκBα S32/S36 Phosphorylation | Non-specific Phosphorylation |
|---|---|---|---|---|---|
| Y169 | DFG+1 | Y169F | Unaffected | Severely compromised | Unaffected |
| K44 | ATP-binding | K44M | Abolished | Abolished | Retained |
| S177/S181 | Activation loop | S177E/S181E | Constitutive | Constitutive | Unaffected |
Experimental Protocols for Studying IKK2 Autophosphorylation
Protocol 1: IKK2 Autophosphorylation Assay
Purpose: To assess IKK2 autophosphorylation at serine and tyrosine residues.
Materials:
- Recombinant full-length IKK2: Purified from appropriate expression system [23] [20]
- Kinase reaction buffer: 25 mM HEPES (pH 7.4), 150 mM NaCl, 10 mM MgCl₂, 1 mM DTT [23]
- ATP solution: 100 μM ATP in reaction buffer [23]
- NEMO protein: Optional, for assessing enhancement of phosphorylation specificity [23]
- SDS-PAGE equipment and phospho-specific antibodies [23]
Procedure:
- Incubate 1 μg of recombinant IKK2 in kinase reaction buffer with 100 μM ATP for 0-60 minutes at 30°C [23].
- Include control reactions without ATP and with kinase-dead IKK2 (K44M) to distinguish specific autophosphorylation [23] [20].
- Stop reactions by adding SDS-PAGE sample buffer and heating to 95°C for 5 minutes [23].
- Resolve proteins by SDS-PAGE and transfer to membranes for immunoblotting [23].
- Probe with phospho-specific antibodies against pS177/pS181 and phosphotyrosine to detect autophosphorylation [23] [20].
- For time-course experiments, collect aliquots at 0, 5, 15, 30, and 60 minutes [23].
Expected Results: Wild-type IKK2 should show time-dependent phosphorylation at both serine and tyrosine residues, producing diffuse slower-migrating bands on SDS-PAGE [23]. The K44M mutant should show abolished phosphorylation, confirming autophosphorylation is self-catalyzed [23] [20]. Addition of NEMO may produce sharper phospho-bands, suggesting enhanced phosphorylation specificity [23].
Protocol 2: IκBα Phosphorylation Assay
Purpose: To evaluate IKK2-mediated phosphorylation of IκBα at specific serines.
Materials:
- Autophosphorylated IKK2: Prepared as in Protocol 1 [20]
- Recombinant IκBα substrate: Full-length or N-terminal fragment containing S32/S36 [23] [20]
- Reaction buffers: As in Protocol 1 [23]
- ATP or ADP solutions: 100 μM each, prepared fresh [20]
- Phospho-specific IκBα antibodies: Specifically recognizing pS32 and pS36 [23]
Procedure:
- Pre-activate IKK2 by incubating with ATP for 30 minutes at 30°C to allow autophosphorylation [23] [20].
- Add 2 μg IκBα substrate to autophosphorylated IKK2 [23].
- Supplement reactions with either: (a) 100 μM ATP (conventional phosphorylation), or (b) 100 μM ADP only (phospho-relay assay) [20].
- Incubate for 30 minutes at 30°C [23] [20].
- Terminate reactions with SDS-PAGE buffer and analyze by immunoblotting [23].
- Probe membranes with phospho-specific IκBα antibodies (pS32, pS36) and total IκBα antibody for normalization [23] [20].
Expected Results: With ATP, wild-type IKK2 should robustly phosphorylate IκBα at S32/S36, while Y169F and K44M mutants show severely impaired phosphorylation [23] [20]. In ADP-only conditions, pre-autophosphorylated IKK2 should mediate phospho-relay to IκBα, demonstrating the unconventional transfer mechanism [20] [24].
Protocol 3: Cellular Validation in Reconstituted MEF Cells
Purpose: To validate IKK2 autophosphorylation mutants in a cellular context.
Materials:
- IKK1/IKK2 double-knockout MEF cells: For clean background [23] [20]
- Expression vectors: Encoding IKK2-WT, IKK2-Y169F, IKK2-K44M [23]
- TNF-α: For pathway stimulation [23] [20]
- Cell lysis buffer: With phosphatase and protease inhibitors [23]
- Immunoprecipitation reagents: For IKK2 or IκBα isolation [23]
Procedure:
- Reconstitute IKK1/IKK2 double-knockout MEF cells with IKK2-WT, IKK2-Y169F, or IKK2-K44M expression vectors [23] [20].
- Culture cells for 24-48 hours to allow protein expression [23].
- Stimulate with TNF-α (10-20 ng/mL) for 0-60 minutes to activate the NF-κB pathway [23] [20].
- Lyse cells in appropriate buffer containing phosphatase and protease inhibitors [23].
- Analyze lysates by SDS-PAGE and immunoblotting for pIκBα (S32/S36), total IκBα, and IKK2 expression [23] [20].
- Optionally, immunoprecipitate IKK2 complexes to assess autophosphorylation status [23].
Expected Results: Cells expressing IKK2-WT should show robust IκBα phosphorylation after TNF-α stimulation, while IKK2-Y169F and IKK2-K44M mutants should display severely impaired phosphorylation, confirming the essential role of these residues in cellular contexts [23] [20].
Table 2: Key Research Reagent Solutions for IKK2 Phosphorylation Studies
| Reagent | Function | Application Examples | Considerations |
|---|---|---|---|
| Recombinant IKK2 (wild-type) | Catalytic core for phosphorylation assays | Autophosphorylation studies; Substrate phosphorylation | Requires proper folding domains (KD, ULD, SDD) for full activity [23] |
| IKK2 mutants (Y169F, K44M) | Mechanistic studies to dissect specific functions | Determining residue-specific contributions; Controls for experiments | K44M loses all specific activity; Y169F specifically affects S32 phosphorylation [23] [20] |
| NEMO/IKKγ | Regulatory scaffold protein | Enhancing phosphorylation specificity in vitro; Complex assembly studies | Sharpens autophosphorylation band pattern on gels [23] |
| Recombinant IκBα | Native substrate for IKK2 | Phosphorylation assays; Specificity studies | Must contain N-terminal domain (S32/S36) for signal-responsive phosphorylation [23] [20] |
| Phospho-specific antibodies (pS32/pS36 IκBα) | Detection of specific phosphorylation events | Western blotting; Monitoring pathway activation | Critical for assessing signal-responsive phosphorylation fidelity [23] [20] |
Visualizing IKK2 Signaling and Experimental Workflows
IKK2 Activation and Phospho-Relay Mechanism
Experimental Workflow for IKK2 Phosphorylation Studies
Discussion and Functional Implications
The discovery of IKK2's dual-specificity autophosphorylation represents a fundamental advancement in our understanding of this prototypical kinase [23] [24]. The evidence supporting tyrosine autophosphorylation and the subsequent phospho-relay mechanism to IκBα suggests an unconventional evolutionary adaptation that ensures fidelity in NF-κB activation [20] [24]. This mechanism, involving transfer of phosphate from an IKK2 tyrosine to the substrate IκBα rather than direct transfer from ATP, contrasts with conventional kinase mechanisms and may represent a more widespread regulatory strategy among eukaryotic kinases [20].
The critical dependence of IκBα S32 phosphorylation on Y169 autophosphorylation reveals a previously unknown layer of regulation in the NF-κB signaling pathway [23] [20]. This hierarchical control mechanism, where serine autophosphorylation (S177/S181) enables subsequent tyrosine autophosphorylation (Y169) which in turn enables substrate phosphorylation, provides multiple regulatory checkpoints for signal fidelity [23] [24]. The retention of non-specific phosphorylation activity in IKK2-K44M mutant while losing specific S32/S36 phosphorylation suggests that substrate specificity and catalytic activity are structurally and mechanistically separable in IKK2 [23] [20].
These findings open several promising research directions. First, the structural basis for the phospho-relay mechanism requires elucidation through advanced techniques like cryo-EM. Second, the potential existence of similar mechanisms in other kinases warrants systematic investigation. Third, the Y169 site and the phospho-relay mechanism represent novel therapeutic targets for inflammatory diseases and cancers involving dysregulated NF-κB signaling [23] [24]. Developing compounds that specifically disrupt the phospho-relay without affecting overall kinase activity could achieve more precise pathway modulation than current ATP-competitive inhibitors.
This case study establishes that IKK2's dual-specificity autophosphorylation, particularly at Y169, and its unconventional phospho-relay mechanism are essential features ensuring high-fidelity phosphorylation of IκBα at S32/S36 during NF-κB activation [23] [20] [24]. The experimental protocols outlined provide robust methodologies for investigating this mechanism further, while the visualizations clarify the complex relationships between different phosphorylation events. These findings not only redefine our understanding of IKK2's catalytic mechanism but also highlight the importance of comprehensive kinase characterization beyond canonical activation mechanisms. The integration of biochemical, cellular, and structural approaches will be crucial for fully elucidating the functional significance of these discoveries and leveraging them for therapeutic development.
Protein kinases are fundamental enzymes in eukaryotic cellular signaling, catalyzing the transfer of a phosphate group from ATP to specific serine, threonine, or tyrosine residues on target proteins. This phosphorylation event serves as a molecular switch that profoundly regulates protein function, localization, and activity. The catalytic activity of protein kinases is tightly controlled through complex regulatory mechanisms, with activation loop phosphorylation representing one of the most ubiquitous and critical means of regulation. Understanding these mechanisms is paramount for drug discovery efforts, particularly in oncology, where kinase dysregulation is a primary driver of disease pathogenesis [25] [26].
The kinase domain is characterized by a highly conserved bilobal fold consisting of an N-lobe and a C-lobe, with the active site situated in the cleft between them. ATP binds in this cleft, positioning its γ-phosphate for transfer to the protein substrate. Several key structural elements work in concert to facilitate and regulate catalysis. The glycine-rich loop assists in organizing the γ-phosphate for phosphoryl transfer, while the activation loop (A-loop), a region of high sequence divergence and structural flexibility, plays a pivotal role in substrate binding and recognition. The correct positioning of the αC-helix, maintained by a conserved salt bridge between a lysine in strand β3 and a glutamate in the αC helix, is essential for the catalytic machinery to function properly [25] [27].
Table 1: Core Structural Elements of the Protein Kinase Domain
| Structural Element | Function | Role in Catalysis |
|---|---|---|
| N-lobe | Binds ATP | Orients ATP γ-phosphate for transfer |
| C-lobe | Binds protein substrate | Provides specificity and binding surface |
| Activation Loop (A-loop) | Substrate recognition, regulation | Creates high-affinity substrate binding site when ordered |
| Glycine-rich Loop | ATP coordination | Organizes γ-phosphate of ATP for nucleophilic attack |
| αC-helix | Catalytic spine formation | Positions catalytic glutamate for salt bridge with lysine |
| DFG Motif | Magnesium ion coordination | Positions Mg²⁺ ions for ATP coordination in active state |
Molecular Mechanisms of Kinase Activation
The Role of Activation Loop Phosphorylation
Activation loop phosphorylation serves as a master regulatory switch for a vast majority of protein kinases, particularly those within the RD kinase class. This post-translational modification triggers a disorder-to-order transition that stabilizes the loop in a conformation conducive to catalysis. In its unphosphorylated state, the activation loop is often dynamic and disordered, which can physically block substrate access to the active site or prevent the proper alignment of catalytic residues. Phosphorylation of a conserved residue within this loop, often a threonine, serine, or tyrosine, introduces a doubly negative charge that is stabilized by an extensive network of hydrogen bonds with conserved arginine and histidine residues from both the N-lobe and C-lobe [25] [27].
This phosphorylation-induced stabilization has several critical consequences. First, it docks the activation loop against the surface of the C-lobe, creating a rigid platform for substrate binding. Second, it helps organize the catalytic spine (C-spine) and regulatory spine (R-spine)—hydrophobic ensembles that traverse both lobes of the kinase and are essential for maintaining the active conformation. The R-spine, which includes the DFG phenylalanine, is particularly important as it effectively "zips up" the N- and C-lobes, aligning them for efficient catalysis. The stability imparted by the phosphorylated activation loop is reflected in biochemical assays, which show that mutations preventing this phosphorylation can lead to a 20-fold decrease in the apparent phosphoryl transfer rate, as demonstrated in studies of Protein Kinase A (PKA) [27].
Conformational States and Dynamics
Kinase activation is governed by conformational transitions of two highly dynamic structural elements: the activation loop and the αC-helix. These elements can adopt distinct active and inactive conformations that are in dynamic equilibrium. The active state is characterized by a DFG-in orientation (where the aspartate points into the active site to coordinate magnesium ions) and an αC-in conformation (where the catalytic glutamate forms a salt bridge with a conserved lysine). Inactive states, by contrast, can feature a DFG-out conformation, where the DFG phenylalanine occupies the ATP-binding site, and/or an αC-out conformation, where the salt bridge is broken [26].
The energy barrier between these states can be modulated by various regulatory inputs. Phosphorylation of the activation loop is a primary mechanism that shifts the conformational equilibrium toward the active state. For some kinases, this shift involves large-scale domain movements. For others, like ERK2, phosphorylation induces only small conformational changes yet results in a dramatic 50,000-fold increase in catalytic activity. In ERK2, phosphorylation reorganizes a key salt bridge between Lys52 and Glu69 and promotes the binding of a second magnesium ion, creating a catalytically competent active site without major structural rearrangement [28]. These dynamics can be tracked in solution using advanced biophysical techniques such as intramolecular FRET and double electron-electron resonance (DEER) spectroscopy, which have revealed that many kinase inhibitors work by stabilizing specific conformational states [26].
Experimental Analysis of Kinase Conformation and Activity
Biochemical Assays for Kinase Activity
A diverse array of biochemical assays is available to quantify kinase activity and inhibition, each with distinct advantages and applications in drug discovery. These assays generally fall into two categories: activity assays, which directly measure the catalytic formation of phosphorylated products, and binding assays, which assess the affinity of small molecules for the kinase target.
Table 2: Key Biochemical Assay Technologies for Kinase Research
| Assay Technology | Detection Principle | Key Applications | Advantages/Limitations |
|---|---|---|---|
| Radiometric (SPA) | Measures incorporation of ³³P from ATP into substrate | Broad applicability, no antibody needed | Gold standard; requires radioactive handling |
| Luminescence-based (ADP-Glo) | Detects ADP formation via luminescent signal | High-throughput screening (HTS) | Non-radioactive, highly sensitive |
| Fluorescence Polarization (FP) | Measures change in rotational mobility of fluorescent phosphopeptide upon antibody binding | HTS, inhibition studies | Homogeneous format; susceptible to compound interference |
| Time-Resolved FRET (TR-FRET) | Energy transfer between antibody-bound lanthanide chelate and acceptor on phosphorylated peptide | HTS, cellular signaling studies | Low background, ratiometric; requires specific antibodies |
| Mobility Shift | Electrophoretic separation of phosphorylated/non-phosphorylated substrates | Kinetic studies, substrate profiling | Direct measurement; lower throughput |
The selection of an appropriate assay depends on the required sensitivity, throughput, and the specific kinase-inhibitor mechanism under investigation. For instance, TR-FRET and FP assays are well-suited for high-throughput screening due to their homogeneous ("mix-and-read") formats, while mobility shift assays provide a direct, non-antibody-dependent quantitative readout of the phosphorylation reaction [1] [29].
Tracking Conformational Changes
Understanding kinase activation requires more than measuring activity; it demands direct observation of conformational dynamics. Intramolecular Förster Resonance Energy Transfer (FRET) has emerged as a powerful technique for this purpose. In a typical FRET sensor construct, a donor fluorophore (e.g., Alexa 488) is attached to the dynamic activation loop, and an acceptor fluorophore (e.g., Alexa 568) is placed on a relatively static region of the kinase domain. Conformational changes that alter the distance between these fluorophores result in measurable changes in FRET efficiency [26].
Quantitative analysis of these changes is best achieved through nanosecond time-resolved fluorescence lifetime spectroscopy, which fits donor fluorescence decay data to model FRET distances. This approach can resolve active and inactive A-loop conformations and quantify population shifts driven by ligand binding. For example, this method was used to screen a panel of 24 ATP-competitive inhibitors against Aurora A kinase, revealing that most inhibitors triggered substantial structural rearrangements and that stabilization of the DFG-in state ranged from 0.5 to 2.0 kcal/mol [26].
Protocol 1: FRET-Based Analysis of Kinase Conformational Dynamics
- Objective: To quantify activation loop conformational changes in response to ligand binding or phosphorylation.
- Materials:
- Purified kinase, site-specifically labeled with donor (Alexa 488) and acceptor (Alexa 568) fluorophores.
- Ligands/inhibitors of interest.
- Time-resolved fluorescence spectrometer.
- Assay buffer (e.g., 50 mM HEPES pH 7.5, 10 mM MgCl₂, 1 mM DTT).
- Procedure:
- Sample Preparation: Incubate the labeled kinase (10-100 nM) in assay buffer with or without the test ligand for 30 minutes at room temperature.
- Data Acquisition: Transfer samples to a quartz cuvette. Use a pulsed laser to excite the donor fluorophore at 488 nm and record the fluorescence decay of the donor emission at 520 nm with sub-nanosecond time resolution.
- Data Analysis:
- Fit the fluorescence decay data to a model comprising a Gaussian distribution of FRET distances.
- Alternatively, fit to a two-state model to determine the population shift between structural states.
- Calculate the energy transfer efficiency (E) and the inter-dye distance (r) using the formula: E = 1 / [1 + (r/R₀)⁶], where R₀ is the Förster radius of the dye pair.
- Interpretation: A decrease in FRET efficiency/distance indicates a movement of the activation loop away from the reference point, often associated with a DFG-in/active state. An increase suggests a movement toward the reference point, often associated with a DFG-out/inactive state [26].
For higher-resolution structural information, Double Electron-Electron Resonance (DEER), a pulsed electron paramagnetic resonance technique, can be employed. While lower in throughput, DEER provides unambiguous assignment of conformational states and can quantify population shifts by measuring distances between site-specifically introduced spin labels [26].
Research Reagent Solutions for Kinase Studies
A successful kinase research program relies on a toolkit of high-quality reagents and assays. The table below summarizes essential materials and their applications.
Table 3: Essential Research Reagent Solutions for Kinase Activation Studies
| Reagent / Material | Function | Example Application |
|---|---|---|
| Baculovirus/Sf9 Insect Cell System | High-yield expression of recombinant, post-translationally modified kinases | Production of phosphorylated, active kinases for structural and biochemical studies |
| Site-Directed Mutagenesis Kits | Generation of phospho-dead (Ala), phospho-mimetic (Asp/Glu), and catalytic-dead (Asp-Asn) mutants | Mechanistic studies to dissect the role of specific residues in activation and catalysis [25] |
| TR-FRET Kinase Assay Kits | Homogeneous, antibody-based detection of phosphopeptides | High-throughput screening of compound libraries for kinase inhibitors |
| HDX-MS Reagents & Platform | Hydrogen-Deuterium Exchange Mass Spectrometry for probing protein dynamics and conformational changes | Mapping conformational changes and allosteric communication upon phosphorylation or ligand binding [30] |
| Covalent Probe Kits (e.g., for BTK, EGFR) | Irreversible inhibitors that bind to non-catalytic cysteines | Target engagement studies in cells, assessment of pharmacodynamic effects |
| Phospho-Specific Antibodies | Immunodetection of phosphorylated activation loops (e.g., pT197-PKA, pY402-Pyk2) | Monitoring kinase activation status in cellular lysates or tissue samples via Western blot |
Kinase Signaling Cascades and Therapeutic Targeting
Signaling Cascades in Physiology and Disease
Kinases rarely function in isolation; they operate within elaborate signaling cascades such as the Mitogen-Activated Protein Kinase (MAPK) and PI3K/AKT/mTOR pathways. These cascades relay signals from cell surface receptors to intracellular targets, ultimately regulating gene expression, cell proliferation, and survival. In the MAPK cascade, a signal is transmitted via a series of phosphorylation events: Ras activates Raf, which phosphorylates and activates MEK, which in turn phosphorylates and activates ERK. This multi-tiered architecture allows for signal amplification and the integration of multiple regulatory inputs [31].
A critical feature of these pathways is their frequent dysregulation in human diseases, especially cancer. Oncogenic mutations often perturb the conformational equilibrium of kinases, destabilizing inactive states or stabilizing active states. For example, the V600E mutation in BRAF and the L858R mutation in EGFR are thought to destabilize the inactive αC-out conformation, leading to constitutive kinase activity and uncontrolled cell growth [26] [31].
Diagram 1: The MAP Kinase (MAPK) Signaling Cascade. This simplified view shows a classic phosphorylation relay that transduces signals from the cell surface to the nucleus.
Therapeutic Inhibition and Conformation-Selective Drugs
The deep understanding of kinase activation mechanisms has been instrumental in developing targeted cancer therapies. Small-molecule kinase inhibitors are classified based on their binding mode and the conformational state they stabilize. Type I inhibitors bind the active, DFG-in conformation of the kinase. Type II inhibitors extend into a hydrophobic backpocket that is only present in the DFG-out conformation, while Type 1.5 inhibitors recognize the αC-out conformation [26].
A significant advancement in this field is the rise of Targeted Covalent Inhibitors (TCIs). These drugs, such as osimertinib (EGFR) and ibrutinib (BTK), contain a reactive group (often an acrylamide) that forms a covalent bond with a cysteine residue near the ATP-binding site. This mechanism provides prolonged target inhibition and can overcome resistance to reversible inhibitors. As of 2025, eleven orally effective kinase TCIs have been approved by the FDA for indications ranging from non-small cell lung cancer to alopecia areata [32].
Protocol 2: HDX-MS for Mapping Phosphorylation-Induced Conformational Changes
- Objective: To identify regions of a kinase that undergo changes in dynamics and solvent accessibility upon activation loop phosphorylation.
- Materials:
- Purified unphosphorylated and phosphorylated kinase (≥ 5 µg per sample).
- Deuterated buffer (e.g., D₂O-based PBS, pD 7.4).
- Liquid handling robot for precise timing.
- UPLC system with pepsin column online coupled to a mass spectrometer.
- Quench solution (low pH, low temperature).
- Procedure:
- Dilution and Labeling: Dilute the kinase (phosphorylated and unphosphorylated) into deuterated buffer to initiate labeling. Incubate for multiple time points (e.g., 10 s, 1 min, 10 min, 1 h).
- Quenching: After each time point, transfer the reaction to a low-pH quench buffer to reduce pH to ~2.5 and temperature to 0°C, effectively stopping the exchange.
- Digestion and Analysis: Inject the quenched sample onto an immobilized pepsin column for rapid digestion. Separate the resulting peptides by UPLC and analyze by high-resolution mass spectrometry.
- Data Processing: Use specialized software to identify peptides and calculate deuterium incorporation for each peptide at each time point.
- Interpretation: A decrease in deuterium incorporation in the phosphorylated state indicates reduced solvent accessibility, suggesting that region has become more structured or protected (e.g., the activation loop upon phosphorylation). An increase suggests increased dynamics or unfolding. This technique was key in showing that nucleotide engagement stabilizes the autoinhibitory interface in Pyk2, while phosphorylation deprotects regulatory surfaces [30].
The activation of protein kinases through activation loop phosphorylation and associated conformational changes represents a fundamental paradigm in cellular signaling. The transition from an inactive to an active state involves precise structural rearrangements that align catalytic residues, stabilize hydrophobic spines, and create competent substrate-binding surfaces. Contemporary research tools, ranging from FRET and HDX-MS for analyzing dynamics to TR-FRET and biochemical assays for measuring activity, provide a comprehensive toolkit to dissect these mechanisms. This deep mechanistic understanding, particularly of the conformational landscapes that kinases navigate, continues to drive the development of novel therapeutic agents, including conformation-selective and covalent inhibitors, offering profound benefits in the treatment of cancer and other diseases driven by aberrant kinase signaling.
Essential Cofactors and Buffer Conditions for Kinase Activity
Protein kinases are fundamental regulators of cellular processes, controlling growth, differentiation, and metabolism through enzymatic phosphate transfer to protein substrates [29]. The vast majority of signaling pathways involve reversible protein phosphorylation, with dynamics that provide critical insights into how signaling networks function and interact [33]. Assessing kinase activity requires carefully optimized experimental conditions that preserve enzymatic function and enable accurate detection of phosphorylation events. This application note provides a comprehensive framework for establishing robust kinase assay systems, detailing essential cofactors, optimized buffer conditions, and practical protocols for researchers and drug development professionals engaged in kinase research and inhibitor screening.
Essential Cofactors for Kinase Reactions
Kinase enzymes require specific cofactors to facilitate the transfer of phosphate groups from ATP to protein substrates. These cofactors are indispensable for proper enzymatic function and must be carefully controlled in experimental settings.
Table 1: Essential Cofactors for Kinase Activity
| Cofactor | Function | Typical Concentration Range | Notes |
|---|---|---|---|
| ATP | Phosphate group donor | Variable (µM to mM) | Concentration must be optimized for each kinase; located in γ position for transfer [29] |
| Magnesium (Mg²⁺) | Primary catalytic cation | 1-20 mM | Most commonly required divalent cation [29] |
| Manganese (Mn²⁺) | Alternative cation | 1-10 mM | Can substitute for Mg²⁺ in some kinase systems [29] |
| Protein/Peptide Substrate | Phosphate group acceptor | Variable | Specificity varies by kinase; can be general or specialized [29] |
The availability of appropriate substrates is crucial for kinase assays. Universal biotinylated substrates like poly-Glutamine-Tyrosine (polyEY) serve well for tyrosine kinases, while generalized substrates such as myosin basic protein or casein, or specialized peptide substrates are employed for serine-threonine kinases [29]. The choice between protein or peptide substrates depends on the specific research question and whether the assay aims to mimic physiological conditions or maximize throughput and consistency.
Buffer Conditions and Optimization
Kinase buffer composition significantly impacts enzymatic efficiency and stability. A properly formulated buffer maintains optimal pH, ionic strength, and reducing conditions while preventing non-specific interactions.
Table 2: Optimized Buffer Components for Kinase Assays
| Component | Purpose | Recommended Concentration | Optimization Tips |
|---|---|---|---|
| Tris-HCl or HEPES | pH Maintenance | 20-50 mM, pH 7.2-7.5 | Maintain physiological pH; HEPES offers better buffering capacity |
| Sodium Chloride (NaCl) | Ionic Strength | 50-150 mM | Modulate to mimic physiological conditions; avoid excessive concentrations |
| Dithiothreitol (DTT) | Reducing Agent | 0.5-1 mM | Prevents oxidation of cysteine residues; prepare fresh |
| BSA | Stabilizing Protein | 0.1-1 mg/mL | Reduces non-specific binding and surface adsorption |
| Detergents (e.g., Tween-20) | Prevent Aggregation | 0.01-0.1% | Minimizes non-specific interactions; critical in solid-phase assays |
The order of addition during reagent preparation can influence assay performance. It is generally recommended to prepare a master mix containing buffer, cofactors, and substrate to which the kinase is added last to initiate the reaction. Key optimization parameters include buffer pH (typically 7.2-7.5), ionic strength, timing, stopping conditions, order of addition, plate type, and assay volume [29]. Systematic optimization of these variables ensures robust and reproducible results across experimental replicates.
Kinase Signaling Pathway
The diagram below illustrates a generalized protein kinase signaling pathway, showing key components and regulatory mechanisms that govern kinase activity in cellular systems.
This pathway illustrates how extracellular signals activate intracellular kinase cascades through receptor binding and second messenger generation, ultimately leading to phosphorylation of transcription factors and specific cellular responses. Phosphatases provide crucial negative feedback regulation by dephosphorylating activated components [29] [34]. The balance between kinase and phosphatase activity determines the net phosphorylation state of cellular proteins and thus the ultimate biological outcome.
Experimental Protocols
pIMAGO-Based Phosphoprotein Detection in Western Blot Format
The pIMAGO technology provides a non-antibody-based approach for universal phosphorylation detection, utilizing water-soluble, globular nanopolymers multi-functionalized with Ti(IV) ions for phosphate group recognition and reporting groups for detection [33].
Materials:
- pIMAGO reagent (Tymora Analytical, cat. no. PMGO)
- Avidin-peroxidase conjugate (Sigma, cat. no. A3151) or avidin-fluorophore conjugate
- Blocking buffer: 1-5% BSA in TBST
- pIMAGO buffer: 50 mM HEPES, 150 mM NaCl, 0.1% Tween-20, pH 7.4
- Washing buffer: 50 mM HEPES, 150 mM NaCl, 10 mM EDTA, 0.1% Tween-20, pH 7.4
- 1× TBST: 20 mM Tris, 150 mM NaCl, 0.1% Tween-20, pH 7.4
Procedure:
- Sample Preparation: Boil samples for 5 minutes in 1× LDS/SDS sample loading buffer with 20 mM fresh dithiothreitol. Cool to room temperature and add 400 mM iodoacetamide to 80 mM final concentration. Incubate in dark for 15 minutes [33].
- SDS-PAGE and Transfer: Load samples onto gel alongside phosphorylated protein control (10-100 ng). Run SDS-PAGE and transfer to PVDF or nitrocellulose membrane using Tris-glycine transfer buffer for lowest background. For contamination reduction, include second PVDF membrane before the gel in transfer stack [33].
- Phosphoprotein Detection:
- Block membrane with Blocking buffer for 1 hour with shaking (or overnight at 4°C).
- Prepare 1:1,000 pIMAGO reagent in pIMAGO buffer. Incubate with membrane for 1 hour with shaking.
- Wash membrane 3 times with Washing buffer and once with 1× TBST (5 min each).
- Prepare 1:1,000 avidin-peroxidase/fluorophore in Blocking buffer. Incubate with membrane for 1 hour.
- Wash membrane 3 times with 1× TBST (5 min each).
- Detect signal using fluorescence scanner or chemiluminescence substrate [33].
pIMAGO-Based Phosphoprotein Detection in ELISA Format
This higher-throughput approach enables screening of multiple samples simultaneously, ideal for kinase activity profiling and inhibitor screening.
Materials:
- 96-well clear High Bind polystyrene plate (Sigma, cat. no. CLS3590)
- Carbonate buffer: 50 mM carbonate-bicarbonate, pH 9.6
- Blocking buffer for microplate: 1-5% BSA in PBS
- Colorimetric TMB peroxidase substrate kit (Bio-Rad, cat. no. 172-1066)
Procedure:
- Protein Immobilization: Coat wells with protein sample in carbonate buffer overnight at 4°C or 2 hours at room temperature.
- Blocking: Block plates with Blocking buffer for 1-2 hours.
- Phosphorylation Detection:
- Prepare 1:1,000 pIMAGO reagent in pIMAGO buffer. Add to wells and incubate 1 hour.
- Wash 3 times with Washing buffer and once with 1× TBST.
- Prepare 1:1,000 avidin-peroxidase in Blocking buffer. Add to wells and incubate 1 hour.
- Wash 3 times with 1× TBST.
- Add TMB substrate according to manufacturer instructions. Measure absorbance [33].
ADP-Glo Kinase Assay Protocol
The ADP-Glo assay provides a universal, homogeneous, high-throughput screening method to measure kinase activity by quantifying ADP production during kinase reactions, compatible with up to 1 mM ATP.
Procedure:
- Kinase Reaction: Set up kinase reaction in multiwell plate with volume as low as 5 μL. Include appropriate controls.
- ATP Depletion: Add equal volume of ADP-Glo Reagent to terminate kinase reaction and deplete remaining ATP. Incubate for 40-60 minutes.
- ADP Detection: Add Kinase Detection Reagent to convert ADP to ATP and measure newly synthesized ATP using luciferase/luciferin reaction.
- Measurement: Quantify luminescence using a luminometer. Correlate luminescence to ADP concentration using ATP-to-ADP conversion curve [35].
Kinase Assay Workflow
The following diagram outlines the general workflow for conducting kinase activity assays, from experimental setup to detection and data analysis.
This workflow highlights the critical decision points in kinase assay design, particularly the selection of an appropriate detection method based on research objectives. Western blotting with pIMAGO or antibodies provides specific phosphorylation detection, ELISA formats enable higher throughput screening, while ADP detection methods like ADP-Glo offer homogeneous assay compatibility [33] [35].
Research Reagent Solutions
Table 3: Essential Research Reagents for Kinase Studies
| Reagent | Supplier Examples | Application | Key Features |
|---|---|---|---|
| pIMAGO Reagent | Tymora Analytical (cat. no. PMGO) | Universal phosphoprotein detection | Non-antibody, Ti(IV)-based phosphate recognition, works in Western Blot and ELISA [33] |
| Avidin-Peroxidase Conjugate | Sigma (cat. no. A3151) | pIMAGO detection | Signal generation for chemiluminescent detection [33] |
| ADP-Glo Kinase Assay | Promega | Universal kinase activity screening | Luminescent ADP detection, works with up to 1mM ATP, homogeneous format [35] |
| Phospho-Specific Antibodies | Multiple suppliers | Targeted phosphosite detection | Site-specific, various applications (Western, ELISA, flow cytometry) [9] |
| High Bind Polystyrene Plates | Sigma (cat. no. CLS3590) | ELISA assays | Optimal protein immobilization for solid-phase assays [33] |
| Universal Kinase Activity Kit | R&D Systems | ADP-generating kinase assays | Non-radioactive, quantifies kinase activity via ADP production [9] |
Successful kinase activity analysis requires careful attention to cofactor requirements, buffer optimization, and appropriate detection methodology. The essential cofactors ATP, Mg²⁺, and specific substrates must be provided in properly balanced concentrations within optimized buffer systems to maintain enzymatic activity and specificity. The protocols described here, including pIMAGO-based detection and ADP-Glo assays, provide researchers with robust tools for investigating kinase function across various applications from basic research to high-throughput drug screening. As kinase research continues to evolve, these fundamental cofactor and buffer considerations remain essential for generating reliable, reproducible data in biochemical assays for kinase activity and phosphorylation detection research.
Methodologies in Practice: From Classic Techniques to Cutting-Edge Biosensors
In the field of kinase activity and phosphorylation detection research, the accurate measurement of enzymatic activity is foundational. Kinases, enzymes that catalyze the transfer of a phosphate group from adenosine triphosphate (ATP) to specific substrates, are pivotal regulators of cellular signaling pathways, and their dysregulation is linked to numerous diseases, including cancer and metabolic disorders [1]. Among the various techniques available, radioactive assays utilizing [γ-32P]ATP remain the gold standard for direct, quantitative assessment of kinase function [36] [13]. These assays provide unparalleled sensitivity and reliability, forming the cornerstone of target validation and drug discovery efforts. This application note details the methodologies and protocols for conducting kinase activity assays using both the traditional [γ-32P]ATP filter-binding method and the Scintillation Proximity Assay (SPA), a high-throughput adaptation.
Key Research Reagent Solutions
The following table catalogues essential reagents and materials required for implementing radioactive kinase assays.
Table 1: Key Research Reagents for Radioactive Kinase Assays
| Reagent/Material | Function/Description |
|---|---|
| [γ-32P] ATP | The radiolabeled co-substrate; provides the radioactive phosphate group (32P) transferred to the substrate during the kinase reaction [36]. |
| Kinase of Interest | The enzyme being studied; often immunoprecipitated from cell lysates or obtained as a purified recombinant protein [36]. |
| Kinase-Specific Substrate | A peptide or protein that contains the consensus phosphorylation sequence for the kinase under investigation (e.g., Kemptide for PKA) [37]. |
| Scintillation Proximity Beads | Microspheres impregnated with scintillant; used in SPA to capture the radiolabeled product and emit light without a separation step [38]. |
| ATP and Mg2+ | Essential co-factors required for the catalytic reaction of the kinase [37]. |
| Lysis/IP Buffer | For tissue homogenization and subsequent immunoprecipitation of the kinase from complex biological samples [36]. |
Established Methodologies and Protocols
The [γ-32P]ATP Kinase Assay Protocol
The in vitro [γ-32P]ATP kinase assay is a definitive method for measuring kinase activity, as it directly quantifies the incorporation of radioactive phosphate into a substrate [36]. The workflow below outlines the core steps of this protocol.
Detailed Experimental Steps:
- Kinase Immunoprecipitation: Isolate the kinase of interest from cell or tissue lysates using a specific antibody. For instance, muscle biopsies were homogenized in RIPA buffer (50 mmol/l Tris·HCl pH 7.5, 50 mmol/l NaF, 500 mmol/l NaCl, 1% Triton X-100, etc.) followed by immunoprecipitation to obtain the kinase for the assay [36].
- Reaction Setup: Combine the immunoprecipitated kinase with a reaction mixture containing:
- Kinase Reaction: Incubate the reaction mix at 30°C or 37°C for a predetermined time (e.g., 10-30 minutes) to allow phosphorylation to proceed linearly [36].
- Reaction Termination: Stop the phosphorylation by adding a strong acid or denaturing agent, such as trichloroacetic acid (TCA) or formic acid.
- Product Separation and Quantification:
- Spot the terminated reaction onto a phosphocellulose filter membrane, which binds the phosphorylated peptide due to its negative charge.
- Wash the membrane extensively with phosphoric acid or water to remove unincorporated [γ-32P]ATP.
- Place the dried membrane in scintillation fluid and measure the incorporated radioactivity using a liquid scintillation counter [36] [13].
Scintillation Proximity Assay (SPA) Protocol
SPA modernizes the radiometric approach by eliminating the need for separation steps, making it ideal for high-throughput screening (HTS) [38]. The assay principle relies on beads that capture the radioactive product and emit a detectable signal.
Detailed Experimental Steps:
- Bead Preparation: Use SPA beads coated with a capture agent, such as streptavidin, for use with a biotinylated substrate, or protein A for antibody-based capture.
- Kinase Reaction: In a microplate well, combine the kinase, a biotinylated substrate, and [γ-33P]ATP (33P is often preferred over 32P for SPA due to its longer half-life and lower energy emission). The HotSpot assay format is a commercial example of this approach [13].
- Product Capture: After the kinase reaction, add the SPA beads. The biotinylated, now radiolabeled, phosphorylated substrate binds to the streptavidin on the beads.
- Signal Detection: The close proximity of the 33P isotope to the scintillant within the bead excites the scintillant, emitting light. Unincorporated [γ-33P]ATP remains free in solution and is too distant to excite the scintillant, thus not contributing to the signal. The microplate is incubated in the dark and then read using a luminescence counter [13] [38]. This "mix-and-read" homogeneous format is a key advantage for automation.
Comparative Analysis and Data
Radioactive assays are often benchmarked against other common detection technologies. The following table summarizes a comparative analysis based on key performance metrics.
Table 2: Comparison of Kinase Activity Assay Technologies
| Assay Technology | Detection Principle | Throughput | Sensitivity | Key Advantages | Key Limitations |
|---|---|---|---|---|---|
| Radiometric ([γ-32P]ATP) | Direct detection of 32P incorporation via scintillation counting [36]. | Medium | Very High (sub-femtogram) [39] | Gold standard; direct and quantitative; low false-positive rate [36] [13]. | Radioactive waste; safety concerns; limited scalability [40]. |
| Scintillation Proximity (SPA) | Energy transfer from radionuclide (e.g., 33P) to scintillant bead upon binding [38]. | High (HTS-ready) | High | Homogeneous (no separation); amenable to automation; safer than 32P [38]. | Signal can be quenched; specialized beads required. |
| Luminescence (e.g., ADP-Glo) | Coupled enzyme reaction detecting ADP formation via luminescence [1] [40]. | High | High | Non-radioactive; safe; good dynamic range. | Susceptible to interference from luciferase inhibitors [40]. |
| Fluorescence (e.g., TR-FRET, FP) | Fluorescence shift upon phosphorylation or binding event [1]. | High | High | Non-radioactive; HTS compatible. | Can be affected by compound interference (fluorescence) [1] [40]. |
| Mobility Shift (e.g., KiMSA) | Electrophoretic separation of phosphorylated vs. non-phosphorylated fluorescent peptide [37]. | Low to Medium | High | Non-radioactive; direct and quantitative. | Lower throughput; not ideal for miniaturized HTS [37]. |
Within the rigorous context of kinase research and drug development, radioactive assays utilizing [γ-32P]ATP continue to provide the benchmark for accuracy and reliability. The direct measurement of phosphate transfer offers quantitative data that is crucial for validating hits in screening campaigns and for mechanistic studies. While non-radioactive methods have advanced significantly, offering excellent throughput and safety, the radiometric filter-binding assay and its HTS-friendly SPA derivative remain the gold standard against which other technologies are measured. Their application is essential for generating high-quality, definitive data in the study of phosphorylation and kinase activity.
The study of kinase activity is a cornerstone of biochemical research, particularly in the context of cellular signaling and drug discovery. Traditional methods for detecting kinase activity have often relied on radiometric assays, which, while considered a gold standard due to their direct measurement and applicability to a wide range of kinases, come with significant drawbacks including hazardous waste and low throughput [41] [42]. The evolution of assay technology has therefore shifted toward non-radiometric universal assays that enable safer, higher-throughput screening while maintaining robust data quality. These advanced systems primarily function by detecting adenosine diphosphate (ADP), a universal byproduct of kinase-mediated phosphorylation, using homogeneous, mix-and-read formats that eliminate the need for multi-step washing procedures and facilitate automation [41].
Universal assay platforms, such as BellBrook Labs’ Transcreener platform, leverage this principle by employing highly specific antibodies to detect nucleotide products like ADP, AMP, GDP, or UDP. This approach provides a single detection chemistry applicable across diverse enzyme families, including kinases, ATPases, GTPases, glycosyltransferases, and methyltransferases [41]. The primary advantage of this universality is the dramatic reduction in assay development time and variability, allowing research and drug development teams to accelerate hit discovery and focus resources on validating true inhibitory compounds. Furthermore, the adaptability of these assays to various readout modes—including fluorescence polarization (FP), fluorescence intensity (FI), and time-resolved FRET (TR-FRET)—ensures compatibility with standard multimode plate readers used in high-throughput screening (HTS) environments [41].
Key Assay Formats and Comparative Analysis
Non-radiometric assays for kinase activity and ADP detection can be broadly categorized into several formats, each with distinct mechanisms, advantages, and limitations. Understanding these differences is critical for selecting the appropriate assay for a specific research context, whether for primary high-throughput screening (HTS), hit validation, or mechanistic studies.
Luminescence-based assays, such as the ADP-Glo assay, provide an indirect but highly sensitive measure of kinase activity by detecting the production of ADP via a luminescent signal. This format is particularly valued for its rapid and economical workflow, making it well-suited for preliminary high-throughput screening where efficiency is paramount. The ADP-Glo assay can process over 300 10-point IC₅₀ curves per day, enabling researchers to obtain preliminary results on kinase activity rapidly. However, a limitation of this system is that it may be suboptimal for testing kinases with very low enzymatic activity and may require counter-screening for compounds that interfere with its detection system [42].
Fluorescence-based immunoassays, such as the Transcreener platform, directly detect ADP using competitive immunoassays with fluorescent tracers. These assays are homogeneous, mix-and-read formats that require no coupling enzymes or wash steps, minimizing the risk of false positives arising from the inhibition of coupling enzymes. The Transcreener ADP² Assay, for example, consistently achieves robust Z′ factors > 0.7 in 384- and 1536-well plates, indicating excellent screening quality and reliability for HTS. Its direct detection mechanism makes it broadly applicable and less prone to interference compared to coupled enzyme systems [41].
Coupled enzyme assays represent another major category, which rely on a secondary enzymatic reaction to generate a detectable signal. A prominent example is the digital cascade assay implemented in a femtoliter reactor array device (FRAD) for highly sensitive, single-molecule analysis of ATPases and kinases. This assay uses an ADP-triggered cascade reaction that ultimately produces a fluorescent dye, resorufin. The cascade involves ADP-dependent glucokinase (PfGK), glucose-6-phosphate dehydrogenase (G6PDH), and diaphorase, converting ADP into a quantifiable fluorescent signal. This platform allows for the quantification of ADP-producing activities at the single-enzyme level, providing unparalleled sensitivity and the ability to study functional heterogeneity among individual enzyme molecules [43].
Enzyme-coupled fluorometric sensing can also be configured for the simultaneous detection of nanomolar concentrations of ATP, ADP, AMP, adenosine, inosine, and pyrophosphate in extracellular fluids. This multi-analyte profiling is crucial for understanding purinergic signaling cascades and the complex patterns of nucleotide turnover on cell surfaces. These sensing techniques employ a combination of bioluminescent and fluorometric measurements without requiring additional manipulation of sampled biological fluids, thus providing a comprehensive view of purine homeostasis in intact tissues and various cell types [44].
For a clear comparison of the primary non-radiometric assay formats, the table below summarizes their key attributes:
Table 1: Comparison of Key Non-Radiometric Universal Assay Formats
| Assay Format | Detection Mode | Pros | Cons | Ideal Use Cases |
|---|---|---|---|---|
| Luminescence (e.g., ADP-Glo) | Luminescent detection of ADP | Fast, economical, scalable for HTS | May need counterscreens for compound interference; suboptimal for very low-activity kinases | Preliminary high-throughput screening, rapid activity assessment [42] |
| Fluorescence Immunoassay (e.g., Transcreener) | Fluorescence Polarization (FP), Intensity (FI), or TR-FRET | Homogeneous (mix-and-read), low interference, robust for HTS (Z' > 0.7), broad applicability | Limited to nucleotide-producing enzymes | Primary HTS, profiling across kinase, ATPase, GTPase targets [41] |
| Coupled Enzyme / Digital Cascade | Fluorometric detection via enzyme cascade (e.g., resorufin) | Single-molecule sensitivity, quantifies molecule-to-molecule heterogeneity | Complex reagent setup, lower throughput | Sensitive biomarker detection, single-enzyme kinetic studies, functional heterogeneity analysis [43] |
| Enzyme-coupled Multi-Analyte Sensing | Bioluminescent & Fluorometric | Simultaneous detection of ATP, ADP, AMP, adenosine, etc. | Specialized setup for extracellular fluid analysis | Studying purine homeostasis, ectoenzymatic pathways, purinergic signaling [44] |
Detailed Experimental Protocol: Digital Cascade Assay for ADP-Producing Enzymes
The following protocol describes the implementation of a digital cascade assay for the highly sensitive detection of ADP-producing enzymes, such as kinases, using a femtoliter reactor array device (FRAD) [43]. This method enables quantitative measurement of activity at the single-molecule level.
Principle
The assay is based on an ADP-triggered enzyme cascade that culminates in the production of the fluorescent molecule resorufin. Individual enzyme molecules are stochastically encapsulated in microwells. If an enzyme molecule is present and active, it produces ADP, which initiates the cascade, leading to a fluorescent signal in that specific reactor. Counting the fluorescent reactors allows for absolute quantification of active enzyme molecules.
Materials and Reagents
- Femtoliter Reactor Array Device (FRAD): A microdevice containing an array of ~4 μm diameter wells with a volume of approximately 1 fL [43].
- Assay Buffer A: 50 mM potassium phosphate buffer (pH 7.5), containing 100 mM KCl, 2 mM MgCl₂, and 0.02% Tween 20 [43].
- Cascade Enzyme Cocktail:
- ADP-dependent glucokinase (PfGK) from Pyrococcus furiosus (10 U/mL)
- Glucose-6-phosphate dehydrogenase (G6PDH) from E. coli (1 U/mL)
- Diaphorase (GsDI) from Geobacillus stearothermophilus (1 U/mL)
- Fluorogenic Substrate System:
- 1 mM Glucose
- 100 μM NADP⁺
- 100 μM Resazurin
- Enzyme Solution: The kinase or ATPase of interest, diluted to an appropriate concentration in Assay Buffer A. A typical starting concentration is in the low nanomolar range, requiring optimization to achieve a mean of less than one enzyme molecule per reactor (λ < 1) for single-molecule analysis [43].
- Oil Phases:
- Flush oil: AE3000 containing 0.1% SURFLON S-386
- Sealing oil: Fomblin Y25
- Instruments:
- Fluorescence microscope equipped with a time-lapse imaging system and a suitable filter set for resorufin (Ex/Em ~570/590 nm).
- Microfluidic injection system for loading the FRAD.
Procedure
- Cocktail Preparation: Prepare the working solution by supplementing Assay Buffer A with the cascade enzyme cocktail (PfGK, G6PDH, diaphorase) and the fluorogenic substrate system (glucose, NADP⁺, resazurin). Add 1 mM ATP to initiate the kinase reaction in subsequent steps [43].
- Enzyme Encapsulation:
- Dilute the enzyme solution into the cocktail prepared in Step 1.
- Load the mixture into the flow channel of the FRAD.
- Inject the flush oil to segment the aqueous solution into water-in-oil droplets, each occupying an individual microwell.
- Seal the reactors by injecting the sealing oil to prevent evaporation.
- Incubation and Imaging:
- Incubate the FRAD at the desired temperature (e.g., 25°C or 37°C).
- Acquire time-lapse fluorescence images of the entire reactor array at regular intervals (e.g., every 5 minutes) for the duration of the reaction (e.g., 30-60 minutes).
- Data Analysis:
- Image Processing: Identify positive reactors (those with a fluorescence intensity significantly above the background).
- Quantification: The number of active enzyme molecules is determined by counting the positive reactors. The fraction of positive reactors follows a Poisson distribution, allowing for the calculation of the absolute concentration of active enzyme in the original solution.
- Kinetic Analysis: The fluorescence intensity over time in positive reactors can be used to analyze the kinetics of individual enzyme molecules.
The logical and experimental workflow of this digital cascade assay is visualized in the following diagram:
The Scientist's Toolkit: Essential Research Reagent Solutions
Successful implementation of non-radiometric ADP detection assays requires a set of key reagents and materials. The following table details essential components for setting up these experiments, drawing from the protocols and assay descriptions cited.
Table 2: Essential Reagents and Materials for ADP-Detection Assays
| Item | Function / Description | Example Application / Note |
|---|---|---|
| ADP-Specific Antibody | Core component of competitive immunoassays; binds specifically to ADP, displacing a fluorescent tracer. | Used in Transcreener platform for direct, homogeneous ADP detection [41]. |
| Fluorescent Tracer | Competes with native ADP for antibody binding sites; displacement generates a quantifiable signal. | Signal is inversely proportional to ADP concentration (FP, FI, or TR-FRET readouts) [41]. |
| Cascade Enzymes (PfGK, G6PDH, Diaphorase) | Key components for signal amplification in coupled systems; convert ADP to a fluorescent output. | PfGK phosphorylates glucose using ADP; G6PDH reduces NADP+; Diaphorase reduces resazurin to resorufin [43]. |
| Fluorogenic Substrate (Resazurin) | Non-fluorescent precursor converted to fluorescent resorufin in the final step of the enzyme cascade. | Enables highly sensitive, fluorometric detection of ADP in digital and bulk coupled assays [43]. |
| Femtoliter Reactor Array (FRAD) | Microdevice for single-molecule analysis; partitions reactions into thousands of pL-fL volume wells. | Enables digital bioassays for quantifying absolute numbers of active enzyme molecules [43]. |
| Nucleotide Substrates (ATP) | Native substrate for kinases; concentration can be varied to mimic physiological conditions (µM to mM). | Assays can be run at 1 mM ATP to reflect physiological levels for relevant data [42]. |
| Homogeneous Assay Buffer | Aqueous buffer containing essential cofactors (Mg²⁺) and stabilizers (Tween 20) for kinase reactions. | Provides optimal pH and ionic strength for enzyme activity and signal generation [43]. |
The shift from traditional radiometric methods to non-radiometric universal assays represents a significant advancement in biochemical research and drug discovery. Platforms centered on the detection of ADP through luminescence, fluorescence, or sophisticated coupled enzyme systems offer a powerful combination of universality, safety, and high-throughput capability. These assays, such as the Transcreener immunoassays and the ultrasensitive digital cascade assays, provide researchers with robust tools to accurately profile kinase activities, identify inhibitors with high confidence, and even probe enzymatic behavior at the single-molecule level. By adopting these universal assay formats, scientists can streamline their workflow from initial screening to lead optimization, thereby accelerating the pace of discovery in both basic research and therapeutic development.
Within biochemical assay development for kinase activity and phosphorylation detection, the selection and optimization of detection methodologies are paramount. Antibody-based techniques form the cornerstone of this research, enabling the precise quantification and analysis of proteins, their post-translational modifications, and complex interactions. This application note provides a detailed comparison of three pivotal technologies—Western Blot, Enzyme-Linked Immunosorbent Assay (ELISA), and Time-Resolved Förster Resonance Energy Transfer (TR-FRET)—framed within the context of kinase research. We present structured experimental protocols, key reagent solutions, and quantitative data comparisons to guide researchers and drug development professionals in selecting and implementing the most appropriate assay for their specific needs.
The investigation of protein kinases, which regulate cellular processes by phosphorylating serine, threonine, or tyrosine residues, relies heavily on robust detection assays. Western Blot provides semi-quantitative analysis of protein size and expression, ideal for initial validation steps [45] [14]. ELISA offers quantitative, high-throughput measurement of specific protein or phosphoprotein levels from complex mixtures [46] [47]. TR-FRET combines homogeneous, "mix-and-read" formats with high sensitivity, enabling real-time monitoring of dynamic interactions such as kinase activity or protein dimerization in high-throughput screening environments [48] [49] [50]. The distinct advantages and limitations of these techniques are summarized in the table below.
Table 1: Comparative Analysis of Antibody-Based Detection Methods
| Feature | Western Blot | ELISA | TR-FRET |
|---|---|---|---|
| Quantitative Precision | Limited / Semi-Quantitative | Excellent | Good to Excellent |
| Throughput | Low | High | High (Excellent for HTS) |
| Assay Format | Heterogeneous, multi-step | Heterogeneous (traditional) or Homogeneous | Homogeneous ("mix-and-read") |
| Dynamic Monitoring | No (Endpoint) | Conditional (Endpoint typically) | Yes (Real-time possible) |
| Spatial Resolution | Molecular Weight (SDS-PAGE) | No | No (Solution-based) |
| In Vivo Compatibility | No (Lysates) | No (Lysates) | Conditional (Live-cell compatible formats) |
| Key Advantage | Confirms protein size & specificity | Robust, quantitative, high-throughput | Proximity-based, low background, kinetic data |
| Primary Limitation | Low throughput, variable quantification | Requires high-quality matched antibodies | Requires specialized instrumentation & reagents |
Detailed Protocols for Kinase Research
Protocol 1: Western Blot for Low-Abundance Kinase Detection
Application Note: Optimized for detecting low-expression kinases or their phosphorylated forms, such as Tissue Factor (TF) in activated monocytes, which express ~100-fold fewer molecules than cancer cell lines [45].
- Step 1: Sample Preparation. Lyse cells in RIPA buffer supplemented with protease and phosphatase inhibitors. For kinase activation studies, treat cells (e.g., THP-1 monocytes) with relevant stimuli (e.g., 10 μg/mL LPS for 5 hours) prior to lysis [45].
- Step 2: Electrophoresis and Transfer. Load 20-40 μg of total protein per lane on a 4-12% Bis-Tris gel. Transfer proteins to a PVDF membrane using standard wet or semi-dry transfer systems.
- Step 3: Blocking and Antibody Incubation. Block membrane with 5% BSA in TBST for 1 hour at room temperature. Incubate with primary antibody (e.g., anti-TF ab252918, 1:1000 dilution) in blocking buffer overnight at 4°C [45]. Wash and incubate with fluorophore-conjugated secondary antibody (e.g., IRDye 680RD, 1:10,000) for 1 hour at room temperature [45].
- Step 4: Detection and Analysis. Image the membrane using a fluorescence imaging system. Use β-actin (1:5000) or α-actinin (1:1000) as a loading control [45].
Troubleshooting Tip: For low-abundance targets, sensitivity is critically dependent on blocking conditions, antibody affinity, and the detection method. Validate antibodies using knockout controls (e.g., HAP-1 TF KO cells) to confirm specificity [45].
Protocol 2: TR-FRET Kinase Activity Assay
Application Note: This homogeneous protocol is ideal for high-throughput screening of kinase inhibitors or profiling kinase activity, exemplified for Protein Kinase C (PKC) and tyrosine kinases [49].
- Step 1: Reaction Setup. In a low-volume 384-well plate, combine:
- Step 2: Kinase Reaction. Incubate the plate for 60-90 minutes at room temperature [49].
- Step 3: Detection Mix Addition. Add 10 μL of a detection solution containing:
- Step 4: TR-FRET Measurement. Incubate for 60 minutes at room temperature. Read the plate using a time-resolved fluorescence reader (e.g., CLARIOstar or PHERAstar) with excitation at ~337 nm. Measure emission at 490 nm (Terbium donor) and 520 nm (Fluorescein acceptor) after a 100 μs delay [49]. Calculate the 520 nm/490 nm emission ratio.
Troubleshooting Tip: The ratiometric nature of TR-FRET corrects for well-to-well volume variation and compound interference. A Z'-factor > 0.5 indicates a robust assay for HTS [49].
Protocol 3: Sandwich ELISA for Phospho-Protein Quantification
Application Note: Designed for the absolute quantification of specific phosphorylation events, such as Akt phosphorylation at Ser473, a key node in cell survival signaling pathways [46].
- Step 1: Plate Coating. Utilize a commercial pre-coated plate or coat with a capture antibody specific to the target protein (e.g., Phospho-Akt Ser473) overnight.
- Step 2: Blocking and Sample Incubation. Block the plate with a protein-based buffer (e.g., 1% BSA in PBS) for 1 hour. Add cell lysates and serial dilutions of a standard phospho-protein for calibration. Incubate for 2 hours at room temperature.
- Step 3: Detection Antibody Incubation. After washing, add a detection antibody conjugated to Horseradish Peroxidase (HRP), which is also specific to the phosphorylation site, forming a sandwich complex. Incubate for 1-2 hours [46].
- Step 4: Signal Development and Quantification. Add TMB substrate solution. After sufficient color development, stop the reaction with acid. Measure the absorbance at 450 nm. Generate a standard curve from the calibrators to interpolate the concentration of phospho-protein in unknown samples [46].
Troubleshooting Tip: For optimal performance, use matched antibody pairs that recognize non-overlapping epitopes on the target protein to ensure specific and sensitive sandwich formation [47].
The Scientist's Toolkit: Research Reagent Solutions
Successful assay development relies on critical reagents. The table below outlines essential components and their functions.
Table 2: Key Research Reagents for Antibody-Based Assays
| Reagent / Solution | Function & Importance | Example Product / Composition |
|---|---|---|
| Validated Primary Antibodies | Binds specifically to the target protein or post-translational modification (e.g., phosphorylation). Critical for all three techniques. | Anti-phospho-Akt (Ser473) [46]; Anti-TF ab252918 (clone EPR22548-240) for Western [45] |
| Matched Antibody Pairs | A pair of antibodies binding non-overlapping epitopes for highly specific sandwich assays (ELISA, TR-FRET). | CST Matched Antibody Pairs (e.g., for Total or Phospho-proteins) [47] |
| TR-FRET Lanthanide Donor | Long-lived fluorophore (e.g., Eu, Tb) that minimizes background fluorescence via time-gated detection. | LanthaScreen Tb-labeled antibody [49]; CoraFluor-1-labeled nanobody [50] |
| Fluorescent Tracers / Acceptors | Binds the target or antibody, serving as the FRET acceptor; emission indicates proximity. | JQ1-FITC for BRD4 engagement [50]; SureLight APC [48] |
| Specialized Assay Buffers | Provides optimal pH, ionic strength, and blocking to minimize non-specific binding. | Kinase Buffer with DTT/MnCl₂ [49]; TR-FRET Dilution Buffer with EDTA [49] |
Data Analysis and Interpretation
Quantitative Data from Case Studies
Table 3: Representative Quantitative Data from Assay Applications
| Assay Type | Target / Process | Key Quantitative Result | Implication for Kinase Research |
|---|---|---|---|
| Western Blot (Optimized) | Tissue Factor (TF) in low-expressing cells [45] | Detection of TF in LPS-stimulated PBMCs (~17,000 molecules/cell) vs. BxPC-3 (>350,000 molecules/cell). | Enables study of low-abundance kinases in physiologically relevant models. |
| TR-FRET Kinase Assay | PKCα Enzyme Titration [49] | EC50 = 0.43 ng/mL; Z'-factor = 0.94; Signal-to-Noise = 60. | Confirms assay robustness and suitability for high-throughput inhibitor screening. |
| TR-FRET Protein Quantification | Endogenous BRD4 Degradation [50] | Dose-dependent degradation by dBET6 (PROTAC) measured in 1.5 hours from lysis; Z'-factor = 0.75 in 96-well format. | Facilitates rapid, quantitative profiling of kinase-targeting degraders in high-throughput. |
| TR-FRET Dimerization Assay | EGFR/HER2 Heterodimers [51] | Cetuximab + Trastuzumab reduced heterodimers by 72% in SKOV-3 cells. | Allows quantification of receptor kinase dimerization, a key activation mechanism, in response to therapeutics. |
Signaling Pathway and Experimental Workflow
The following diagram illustrates a generalized workflow for a TR-FRET-based kinase activity assay, showcasing the homogeneous "mix-and-read" format and the principle of proximity-dependent signal generation.
The strategic application of Western Blot, ELISA, and TR-FRET provides a powerful, complementary toolkit for advancing kinase research and drug discovery. Western Blot remains indispensable for initial target validation and specificity checks, especially for low-abundance proteins. ELISA delivers robust, quantitative data for focused studies on specific kinase or phosphoprotein levels. TR-FRET emerges as a superior technology for high-throughput screening, enabling dynamic assessment of kinase activity, target engagement, and complex protein-protein interactions with high sensitivity and efficiency. The choice of technique should be guided by the specific research question, required throughput, and need for quantitative precision, as detailed in the protocols and data within this note.
In the field of kinase activity and phosphorylation detection research, characterizing molecular interactions is fundamental for understanding cellular signaling and developing targeted therapies. Kinases regulate numerous cellular processes by catalyzing the transfer of phosphate groups from adenosine triphosphate (ATP) to specific substrates, controlling cell signaling, growth, and metabolism [1]. Advanced binding assays provide critical insights into these mechanisms by measuring binding affinity, stoichiometry, specificity, and kinetics between kinases and their interaction partners, enabling researchers to identify therapeutic candidates and conduct robust drug screening [52] [1].
Fluorescence Polarization (FP) and Thermal Shift Assays (TSA) represent two powerful techniques in the biochemical toolkit for investigating kinase function. These methods enable researchers to study binding events under equilibrium conditions in solution, providing complementary information about kinase-ligand interactions and structural stability [1] [53]. Due to their sensitivity, reproducibility, and adaptability to high-throughput formats, these assays have become indispensable in both basic kinase research and drug discovery pipelines, particularly for identifying and validating kinase inhibitors [1].
Theoretical Principles
Molecular Binding Fundamentals
The binding between a kinase (P) and its ligand (L) to form a complex (P·L) is described by the reversible reaction: P + L ⇄ P·L, characterized by an association rate (kₐ) and dissociation rate (kd) [52]. At equilibrium, the dissociation constant KD = kd/kₐ = [P]free·[L]free/[P·L] quantifies binding affinity, with lower KD values indicating tighter binding [52]. For quantitative analysis, the binding fraction follows a hyperbolic model: f = [P·L]/[P]total = [L]free/([L]free + KD) [52]. Understanding these parameters is essential for characterizing kinase-inhibitor interactions and interpreting data from FP and TSA experiments.
Fluorescence Polarization Principles
Fluorescence Polarization measures changes in the rotational mobility of a fluorescent ligand upon binding to a larger molecule like a kinase. When a fluorescently labeled molecule is excited with polarized light, the emitted light remains polarized if the molecule rotates slowly (bound state), but becomes depolarized if the molecule rotates quickly (free state) [1]. The key relationship governing FP is the Perrin equation, which connects polarization to rotational correlation time: (1/P - 1/3) = (1/P₀ - 1/3)(1 + 3τ/ρ), where P is measured polarization, P₀ is the fundamental polarization, τ is the fluorescence lifetime, and ρ is the rotational correlation time [52]. Since ρ is directly proportional to molecular volume, binding of a small fluorescent tracer to a larger kinase significantly increases ρ, resulting in higher polarization values.
Thermal Shift Assay Principles
Thermal Shift Assays (also known as Thermofluor or differential scanning fluorimetry) monitor protein thermal stability through temperature-dependent unfolding [53]. The method utilizes environmentally sensitive dyes like SYPRO Orange that fluoresce upon binding to hydrophobic regions exposed during protein denaturation [53]. By measuring fluorescence during a controlled temperature ramp, researchers can determine the protein's melting temperature (Tm), where 50% of the protein is unfolded. Ligand binding typically stabilizes the protein structure, increasing Tm proportionally to binding affinity and concentration [53]. This ΔT_m provides a quantitative measure of binding strength and can be used to determine dissociation constants for kinase-ligand interactions.
Fluorescence Polarization Assay
Workflow and Mechanism
The following diagram illustrates the fundamental mechanism and experimental workflow of Fluorescence Polarization for detecting kinase-ligand binding:
Detailed Protocol
Materials and Reagents:
- Purified kinase protein of interest
- Fluorescently labeled ligand or inhibitor (often ATP-competitive)
- Black, clear-bottom microplates (96- or 384-well)
- FP-compatible plate reader with polarizing filters
- Assay buffer (typically PBS or Tris-based with Mg²⁺ for kinases)
- DMSO for compound dilution
Step-by-Step Procedure:
Sample Preparation:
- Prepare assay buffer containing necessary cofactors (e.g., Mg²⁺, DTT).
- Create a dilution series of the kinase in buffer, typically ranging from nM to μM concentrations.
- Keep the fluorescent tracer concentration constant, ideally near or below the expected K_D value.
Plate Setup:
- Add 50-100 μL of each reaction mixture to microplate wells in triplicate.
- Include controls: blank (buffer only), free tracer (tracer without kinase), and maximum polarization (tracer with saturating kinase).
- For inhibitor screening, pre-incubate kinase with compounds before adding fluorescent tracer.
Measurement:
- Equilibrate plate to assay temperature (typically room temperature or 25°C).
- Read polarization in milliP (mP) units on a plate reader with appropriate filters.
- Integration times of 100-500 ms per well are typical.
Data Collection:
- Record mP values for each well.
- Calculate specific binding by subtracting background from free tracer controls.
Data Analysis:
- Plot mP versus kinase concentration and fit to a binding isotherm: mP = mPmin + (mPmax - mPmin) × [Kinase]^n / (KD + [Kinase]^n)
- For competitive displacement experiments, plot mP versus inhibitor concentration and fit to determine IC₅₀, then convert to K_i using Cheng-Prusoff equation.
Key Reagents and Materials
Table 1: Essential Reagents for Fluorescence Polarization Assays
| Reagent/Material | Function/Purpose | Considerations for Kinase Research |
|---|---|---|
| Fluorescent Kinase Inhibitor Tracer | Binds kinase active site; generates signal | Select based on kinase specificity; common tags: FITC, TAMRA, BODIPY |
| Purified Kinase Protein | Binding target | Requires active, properly folded kinase; concentration range: nM-μM |
| FP-Compatible Microplates | Reaction vessel | Black walls minimize signal crosstalk; clear bottom optional |
| Assay Buffer with Mg²⁺ | Maintains kinase activity and stability | Typically contains 1-10 mM MgCl₂; may include reducing agents |
| Reference Inhibitors | Controls for assay validation | Known potent inhibitors for positive and negative controls |
Thermal Shift Assay
Workflow and Mechanism
The following diagram illustrates the thermal denaturation process and detection principle of Thermal Shift Assays:
Detailed Protocol
Materials and Reagents:
- Purified kinase protein (≥0.1 mg/mL)
- SYPRO Orange dye (5000X concentrate)
- Real-time PCR instrument or thermal cycler with fluorescence detection
- Clear PCR tubes or plates (compatible with thermal cycler)
- Test compounds or ligands in DMSO
- Appropriate buffer (e.g., HEPES or PBS, pH 7.0-7.5)
Step-by-Step Procedure:
Sample Preparation:
- Prepare kinase solution in assay buffer at 0.1-1 mg/mL concentration.
- Dilute SYPRO Orange stock 1:5000 in buffer (final 1X concentration).
- For ligand testing, pre-incubate kinase with compound for 15-30 minutes.
- Include controls: buffer with dye only (background), kinase without ligand (reference T_m).
Reaction Setup:
- Combine 18 μL kinase sample with 2 μL 10X SYPRO Orange (final 1X).
- Alternatively, prepare master mix of kinase and dye, then aliquot.
- For 96-well format, use 20-50 μL final volume per well.
- Centrifuge briefly to remove bubbles.
Thermal Denaturation:
- Program thermal cycler with a gradient from 25°C to 95°C with 1°C increments.
- Set fluorescence acquisition at each temperature step using FRET or SYBR Green filter.
- Hold for 10-30 seconds at each temperature for equilibration.
Data Collection:
- Export raw fluorescence versus temperature data.
- Normalize fluorescence between 0% (initial) and 100% (maximum).
Data Analysis:
- Plot normalized fluorescence versus temperature to generate melting curves.
- Calculate first derivative (dF/dT) to determine T_m as the minimum point.
- Determine ΔT_m between ligand-bound and apo kinase samples.
- For KD determination, measure ΔTm at varying ligand concentrations and fit to: ΔTm = ΔTm,max × [L] / (K_D + [L])
Key Reagents and Materials
Table 2: Essential Reagents for Thermal Shift Assays
| Reagent/Material | Function/Purpose | Considerations for Kinase Research |
|---|---|---|
| SYPRO Orange Dye | Binds hydrophobic patches exposed during unfolding | Standard concentration: 1-5X; minimal effect on protein stability |
| Purified Kinase | Target protein for stability assessment | Quality crucial; avoid aggregated or degraded protein |
| Real-time PCR Instrument | Precise temperature control and fluorescence detection | Requires compatible filters (usually ROX/SYPRO Orange) |
| Thermal-stable Plates/Tubes | Withstand repeated thermal cycling | Clear for optical detection; seal to prevent evaporation |
| Reference Ligands | Controls for stabilization | ATP or known inhibitors for validation |
Data Analysis and Interpretation
Quantitative Parameters
Table 3: Key Quantitative Parameters in Binding Assays
| Parameter | Fluorescence Polarization | Thermal Shift Assay | Biological Significance |
|---|---|---|---|
| Primary Readout | Polarization (mP units) | Melting Temperature (T_m) | Direct measurement of binding or stability |
| Binding Affinity | K_D from saturation binding | KD from ΔTm concentration dependence | Lower K_D indicates tighter binding |
| Signal Range | ΔmP between free and bound | ΔT_m between apo and bound | Larger range improves assay robustness |
| Typical Precision | K_D values within 2-fold | T_m ± 0.5°C | Enables accurate ranking of compounds |
| High-throughput Format | 384- or 1536-well plates | 96- or 384-well plates | Suitable for screening compound libraries |
Troubleshooting Common Issues
Fluorescence Polarization Challenges:
- Low signal window: Optimize tracer concentration; ensure kinase is active and properly folded.
- High background: Include free tracer controls; check for compound autofluorescence.
- Non-specific binding: Add carrier protein (BSA, 0.1 mg/mL) or detergent (Tween-20, 0.01%).
- Plate reader limitations: Verify calibration with reference compounds; ensure proper filter alignment.
Thermal Shift Assay Challenges:
- Poor transition curves: Optimize protein concentration; check dye concentration and mixing.
- Multiple transitions: May indicate protein heterogeneity or domain-specific unfolding.
- Small ΔT_m: Ensure ligand solubility; verify protein functionality and binding.
- Evaporation: Seal plates properly; use mineral oil overlay if necessary.
Applications in Kinase Research and Drug Discovery
Fluorescence Polarization and Thermal Shift Assays serve complementary roles in kinase research. FP is particularly valuable for direct binding measurements and competitive displacement assays, providing precise K_D values for kinase-inhibitor interactions [1]. This makes FP ideal for screening compound libraries and determining inhibitor potency and selectivity. The technique's homogeneous format (no separation steps) and suitability for miniaturization have established it as a workhorse in high-throughput screening campaigns targeting kinase families [1].
Thermal Shift Assays offer unique advantages for studying kinase stability and detecting ligand binding, including for allosteric inhibitors that may not compete with ATP [53]. The ability to monitor structural stability makes TSA particularly valuable for optimizing kinase expression constructs, evaluating buffer conditions, and identifying stabilizers that may enhance crystallization success. In drug discovery, the method's capacity to detect direct target engagement, even for weak binders, complements functional assays and provides mechanistic insights into inhibitor effects on kinase conformation [53].
Both techniques contribute significantly to the characterization of kinase inhibitors across different classes (Type I, II, and allosteric inhibitors) and support the development of targeted therapies for cancer, inflammatory diseases, and neurological disorders where kinase dysregulation plays a pathogenic role [1]. The continuous advancement of these methodologies, including the integration of fluorescent ligands and improved detection technologies, continues to expand their applications in kinase research and accelerate the discovery of novel therapeutic agents [52] [1].
The study of kinase activity and phosphorylation detection is a cornerstone of biochemical research, particularly in the context of drug discovery for diseases like cancer. Traditional methods often rely on radioactive isotopes or antibodies, which can be costly, hazardous, and technically challenging. Recent advancements have introduced innovative biosensor platforms that overcome these limitations, enabling sensitive, real-time, and function-independent analysis of protein pharmacology and cellular metabolites. This application note details two cutting-edge platforms: the PhALC (a representative of structural dynamics response assays using α-complementation) and genetically encoded fluorescent reporters. These technologies are reshaping our approach to studying kinase signaling networks and phosphorylation events, providing researchers with powerful tools for high-throughput screening and mechanistic investigation.
Application Notes
PhALC-Based Structural Dynamics Response (SDR) Assays
The PhALC (Split Protein α-Complementation) platform, exemplified by the Structural Dynamics Response (SDR) assay, represents a significant breakthrough in studying protein-ligand interactions, including kinase inhibitors. This technology leverages the principle that ligand binding induces structural dynamic changes in a target protein, which can be transmitted to a fused sensor protein to generate a quantifiable signal [54].
Mechanism and Advantages: The core innovation involves fusing NanoLuc luciferase (NLuc) or its α-complementation peptide (HiBiT) to either the N- or C-terminus of a target protein. Upon ligand binding, conformational changes alter the bioluminescence output of the sensor, providing a gain-of-signal readout that is function-independent and isothermal [54]. This contrasts with traditional enzymatic assays that typically measure loss of signal (inhibition). A key advantage is the ability to study proteins in crude cellular lysates from gene-edited cells, bypassing the need for purification and enabling research in more physiologically relevant environments [54].
Application in Kinase Research and Drug Discovery: The platform's generality has been demonstrated across diverse enzyme classes, including kinases. It enables the detection of nuanced pharmacological effects, such as cofactor-induced synergy in ligand binding and allosteric modulation [54]. For kinase targets, this allows researchers to distinguish between inhibitors that bind in an ATP-dependent versus ATP-independent manner, revealing critical mechanistic insights into compound action. The technology is particularly suited for high-throughput screening (HTS) of large compound libraries, as it is compatible with low-volume formats and does not require specialized reagents like fluorescently-labeled probes or antibodies [54].
Genetically Encoded Fluorescent Reporters
Genetically encoded fluorescent reporters are engineered proteins designed to monitor biochemical events, such as changes in metabolite concentration or protein-protein interactions, in living cells in real time.
Principle and Design: These biosensors typically incorporate a sensing domain (e.g., a ligand-binding domain or a protein-interaction domain) coupled to one or more fluorescent proteins (FPs). Upon encountering the target analyte or a specific molecular event, a conformational change alters the fluorescence properties (intensity, wavelength, or lifetime) of the FPs [55]. Recent developments include FRET-based biosensors and single FP-based designs.
Monitoring Polyamine Dynamics: A recent innovative example is a genetically encoded fluorescent reporter for cellular polyamines, which are ubiquitous metabolites linked to diseases like cancer and Parkinson's disease [56]. This reporter utilizes the polyamine-responsive ribosomal frameshift motif from the OAZ1 gene to transduce polyamine concentration into a fluorescent signal [56]. This allows for real-time measurement of dynamic changes in polyamine levels in single living cells in response to genetic or pharmacological perturbations. Such a tool is invaluable for investigating polyamine biology and for screening therapies that target this pathway.
Broader Context in Cancer Drug Discovery: Fluorescent and bioluminescent biosensors (e.g., FRET, TR-FRET, BRET, NanoBRET, NanoBiT) have become indispensable in cancer research. They have enabled breakthrough discoveries, such as identifying Celastrol as a novel YAP-TEAD inhibitor via NanoBiT-based screening and using NanoBRET assays to detect RAF dimerization [55]. These technologies facilitate the study of complex signaling pathways, protein-protein interactions, and drug effects directly in living cells.
Quantitative Comparison of Biosensor Platforms
The table below summarizes key performance metrics and characteristics of the featured biosensor platforms, providing a direct comparison for researchers.
Table 1: Quantitative Comparison of Innovative Biosensor Platforms
| Feature | PhALC-SDR Assay [54] | Genetically Encoded Polyamine Reporter [56] |
|---|---|---|
| Core Technology | Bioluminescence; Split-NanoLuc α-complementation | Fluorescence; OAZ1 frameshift motif |
| Detection Mode | Gain-of-signal, ligand-dependent bioluminescence | Real-time fluorescence in live cells |
| Primary Application | Ligand binding, allostery, cofactor dependence | Measurement of cellular polyamine concentrations |
| Throughput | High (suitable for HTS) | Medium to High (compatible with screening) |
| Cellular Context | Purified proteins or cell lysates | Single living cells |
| Key Metric | pSDR50 (ligand potency) | Dynamic concentration changes |
| Demonstrated Sensitivity | pSDR50 values ranging from 5.73 to 8.30 (for PTC124 binding FLuc) | Enables genome-wide CRISPR screens (e.g., identified mitochondrial respiration-polyamine import link) |
Essential Research Reagents and Materials
The following table lists key reagents and materials essential for implementing the PhALC-SDR assay and genetically encoded fluorescent reporters.
Table 2: Research Reagent Solutions for Biosensor Implementation
| Reagent/Material | Function/Description | Example Application |
|---|---|---|
| NanoLuc Luciferase (NLuc) | Intact reporter protein; generates bright, ATP-independent bioluminescence [54] | Core sensor in SDR fusion constructs |
| HiBiT Peptide | 11-amino acid α-peptide for NanoLuc complementation [54] | Fused to target protein for SDR assays with LgBiT |
| LgBiT Protein | Large ω-fragment of NanoLuc for complementation with HiBiT [54] | Added to assays to reconstitute luciferase activity with HiBiT-tagged proteins |
| Furimazine | Cell-permeable substrate for NanoLuc luciferase [54] | Provides bioluminescence readout in SDR assays |
| OAZ1 Frameshift Motif | Polyamine-sensing domain from the OAZ1 gene [56] | Sensing element in the genetically encoded polyamine reporter |
| Fluorescent Protein (e.g., GFP, RFP) | Reporter domain that emits fluorescence upon excitation [56] | Output module in genetically encoded fluorescent reporters |
| Kemptide-FITC | Fluorescent-labeled peptide substrate for PKA [37] | Phosphorylation substrate in non-radioactive kinase mobility shift assays (KiMSA) |
Experimental Protocols
Protocol 1: PhALC-SDR Assay for Ligand Binding Quantification
This protocol describes the steps to quantify ligand binding to a target kinase using a HiBiT-tagged protein in a purified system or cell lysate [54].
Step 1: Biosensor Construct Preparation
- Clone the gene of interest (e.g., a kinase) into an expression vector, creating a fusion with the HiBiT tag at either the N- or C-terminus.
- Express the fusion protein in a suitable system (e.g., bacterial, mammalian) and purify it using standard affinity chromatography protocols, or prepare lysates from cells expressing the construct.
Step 2: SDR Assay Setup
- Prepare a reaction buffer compatible with the target protein and NLuc activity.
- In a white, low-volume assay plate, mix the purified HiBiT-tagged protein or cell lysate with LgBiT protein to reconstitute the NLuc enzyme.
- Add the furimazine substrate to initiate the bioluminescence reaction and measure the baseline signal using a luminescence plate reader.
Step 3: Ligand Titration and Data Acquisition
- Prepare a serial dilution series of the test ligand in DMSO, ensuring the final DMSO concentration is consistent and non-interfering (typically <1%).
- Add the ligand dilutions to the assay wells. Include controls: vehicle-only (DMSO) for maximum signal and a known inhibitor for minimum signal if available.
- Incubate the plate isothermally (e.g., at room temperature for 30 minutes) to allow ligand binding and signal stabilization.
- Measure the bioluminescence signal again.
Step 4: Data Analysis
- Normalize the luminescence readings from ligand-treated wells to the vehicle control (defined as 100%).
- Plot the normalized response (%) against the logarithm of the ligand concentration.
- Fit the dose-response curve using a four-parameter logistic model to determine the pSDR50 (the negative log of the ligand concentration that produces a half-maximal signal response) [54].
Protocol 2: Kinase Mobility Shift Assay (KiMSA) for PKA Activity
This protocol provides a detailed method for a non-radioactive, fluorescence-based assay to measure PKA activity, relevant for phosphorylation detection research [37].
Step 1: Sample Preparation (e.g., Sperm Cell Lysate)
- Incubate cells under desired experimental conditions (e.g., non-capacitating vs. capacitating media for sperm cells).
- Centrifuge cells (10,000 × g, 3 min, RT) and discard supernatant.
- Lyse the cell pellet in an appropriate lysis buffer containing protease and phosphatase inhibitors. Incubate on ice for 30 minutes.
- Clarify the lysate by centrifugation, and use the supernatant as the source of kinase.
Step 2: Kinase Reaction
- Prepare a kinase reaction buffer containing HEPES, MgCl₂, ATP, and the fluorescent substrate Kemptide-FITC [37].
- Combine the cell extract with the kinase reaction buffer.
- Incubate the reaction for 25 minutes at 37°C in the dark.
Step 3: Reaction Termination and Electrophoresis
- Stop the kinase reaction by placing tubes on ice and adding Tween-20.
- Heat the samples at 100°C for 1 minute.
- Load the samples onto an agarose gel and perform electrophoresis.
Step 4: Fluorescence Quantification
- Image the gel using a fluorescence imaging system to visualize the phosphorylated and non-phosphorylated Kemptide-FITC bands.
- Perform densitometry analysis on the bands.
- Calculate PKA activity as the percentage of phosphorylated peptide relative to the total peptide (phosphorylated + non-phosphorylated) or as international units (IU) of normalized activity [37].
Schematic Diagrams
PhALC-SDR Assay Mechanism and Workflow
The following diagram illustrates the molecular mechanism and experimental workflow of the PhALC-SDR biosensor platform.
Genetically Encoded Reporter for Metabolite Sensing
This diagram depicts the working principle of a genetically encoded fluorescent reporter for detecting metabolites like polyamines in live cells.
Within kinase research and drug development, understanding substrate specificity is paramount. Protein kinases catalyze the transfer of phosphate groups from ATP to serine, threonine, or tyrosine residues on their protein substrates, thereby regulating nearly every major biological process, from cell division and signal transduction to apoptosis [2] [57]. Dysregulation of these phosphorylation events is a hallmark of numerous diseases, including cancer, diabetes, and central nervous system disorders, making kinases prominent therapeutic targets [2] [58]. A critical step in deciphering signaling networks and developing targeted therapeutics is the precise identification of kinase-substrate relationships. This article details the primary strategies employed for substrate selection, ranging from the use of generic proteins to sophisticated peptide library-based approaches for novel substrate discovery, providing application notes and detailed protocols for researchers and drug development professionals.
| Strategy | Description | Key Advantages | Key Limitations | Typical Applications |
|---|---|---|---|---|
| Generic Protein Substrates | Uses common, well-characterized proteins (e.g., myelin basic protein) as phosphorylation acceptors. | Simple, inexpensive, and provides a general measure of kinase activity. | Does not reveal native substrate specificity; low physiological relevance. | Initial kinase activity confirmation and purification tracking [9]. |
| In Vitro Kinase Assays with Putative Substrates | Incubates purified kinase with a single, purified putative substrate protein in the presence of ATP. | Considered the gold standard for validating direct kinase-substrate relationships. | Low-throughput, laborious; requires prior knowledge of candidate substrates [2]. | |
| Peptide Library-Based Profiling | Uses diverse collections of peptides to determine the amino acid sequence preferences of a kinase. | High-throughput, defines consensus phosphorylation motifs, can identify novel substrates. | May lack structural context of full-length proteins. | Defining kinase specificity; discovering novel substrates [2] [59]. |
| Genetic Screening | Uses genetic manipulation (e.g., siRNA) in model organisms to identify genes that suppress or mimic a kinase mutant phenotype. | Can identify physiologically relevant substrates in a cellular context. | High false-positive rate; difficult in mammalian cells; relationship is indirect [2]. | |
| Phosphoproteomics | Uses mass spectrometry (LC-MS/MS) to identify and quantify thousands of phosphorylation sites from cell lysates after kinase perturbation. | Global, unbiased identification of phosphorylation sites; high-throughput. | Does not directly establish kinase-substrate links; requires complex data analysis and enrichment [57]. | |
| Protein Interaction-Based Screening | Identifies kinase-interacting proteins via methods like yeast-two-hybrid or affinity purification. | Can identify stable complexes. | Often misses transient kinase-substrate interactions [2]. |
Strategies for Substrate Selection and Identification
Generic Proteins and Defined Putative Substrates
The most straightforward method to assess kinase activity involves using generic protein substrates or specific putative substrates in an in vitro kinase assay. In this approach, the immunoprecipitated or purified kinase of interest is incubated with an exogenous substrate in the presence of ATP. The phosphorylated product is then detected using methods such as radiometric, colorimetric, or fluorometric detection [2] [9]. While this method is a cornerstone for validating direct phosphorylation and is considered a gold standard for confirming kinase-substrate relationships, it is low-throughput. It requires prior knowledge or a hypothesis about a specific substrate and does not readily reveal the kinase's full substrate specificity profile [2].
Peptide Library-Based Approaches
To define the substrate specificity of kinases in a high-throughput manner, peptide library approaches have been developed. These methods allow for the systematic profiling of a kinase's preference for amino acids surrounding the phosphorylation site.
- Oriented Peptide Libraries: This method involves incubating a kinase with a library of peptides that have a fixed amino acid at the phosphorylation site (e.g., Ser) but degenerate sequences in the flanking positions. By comparing the amino acid composition of the phosphorylated peptide pool to the initial library, a consensus phosphorylation motif can be derived. For example, this technique was used to define the distinct substrate motifs for the kinases ATM and DNA-PK [59].
- Peptide Microarrays: These consist of hundreds to thousands of peptides synthesized on a solid-phase chip. The kinase is incubated with the array, and phosphorylation events are detected using autoradiography, fluorescence, or immunoblotting with phospho-specific antibodies. This platform is ideal for rapidly characterizing kinase specificity based on primary sequences and can help identify novel substrates containing the consensus motif [2].
- Phage Display: In this method, a library of peptides with random sequences is displayed on the surface of bacteriophages. The phage library is incubated with the immobilized kinase, and phages displaying peptides that are phosphorylated are isolated. After several rounds of selection and enrichment, the peptide sequences of the binders are determined by DNA sequencing, revealing the kinase's substrate preference [2].
- Proteome-Derived Peptide Libraries: A more recent innovation, this approach uses natural peptide libraries generated by proteolytic digestion of a model proteome (e.g., with trypsin or chymotrypsin). This peptide mixture is then treated with the enzyme of interest, and the cleavage products (e.g., neo-N-terminal peptides for proteases) are enriched and identified by LC-MS/MS. While initially developed for carboxypeptidases, the principle of using a proteome-derived library is a powerful tool for profiling enzyme specificity [60].
Advanced Genomic and Proteomic Strategies
For a global, unbiased discovery of kinase substrates, more complex strategies are employed.
- Genetic Screening: This classical genetic approach involves establishing a phenotype for a kinase mutant and then performing genome-wide screening (e.g., with siRNA libraries) to identify genes that suppress or mimic this phenotype. While it can reveal physiologically relevant pathways, it often yields many false positives as it does not directly demonstrate a phosphorylation event [2].
- Quantitative High-Throughput Phosphoproteomics: This is a powerful mass spectrometry-based method for comprehensively identifying phosphorylation sites. Cells are perturbed (e.g., by inhibiting a kinase), and proteins are digested into peptides. Phosphopeptides are enriched using techniques like Immobilized Metal Ion Affinity Chromatography (IMAC) or Metal Oxide Affinity Chromatography (MOAC) with titanium dioxide (TiO₂) [57]. The enriched peptides are then analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS). To quantify changes in phosphorylation, isobaric tags (e.g., TMT) are often used, allowing for the multiplexing of several samples in a single experiment [57]. This method can identify thousands of phosphorylation sites and, when combined with kinase inhibition, point to potential direct substrates.
Experimental Protocols
Protocol 1: In Vitro Kinase Assay with Putative Substrate and Western Blot Detection
This protocol is used to validate whether a purified kinase phosphorylates a specific putative substrate protein.
Materials:
- Purified, active kinase
- Purified substrate protein
- ATP (e.g., 100 µM)
- Kinase assay buffer (e.g., 25 mM Tris-HCl, pH 7.5, 5 mM beta-glycerophosphate, 2 mM DTT, 0.1 mM Na₃VO₄, 10 mM MgCl₂)
- SDS-PAGE gel and Western blot equipment
- Phospho-specific antibody targeting the predicted phosphorylation site
- Antibody for total substrate protein
Procedure:
- Prepare the reaction mix in a microtube on ice:
- 1 µg substrate protein
- 10-100 ng active kinase
- Kinase assay buffer to a final volume of 25 µL
- Start the reaction by adding ATP to a final concentration of 100 µM.
- Incubate at 30°C for 30 minutes.
- Terminate the reaction by adding 6.25 µL of 5X SDS-PAGE loading buffer and heating at 95°C for 5 minutes.
- Load the entire sample onto an SDS-PAGE gel and perform electrophoresis.
- Transfer the proteins to a PVDF or nitrocellulose membrane.
- Block the membrane with 5% BSA in TBST for 1 hour at room temperature.
- Incubate with the primary phospho-specific antibody (diluted in blocking buffer) overnight at 4°C.
- Wash the membrane 3 times for 5 minutes with TBST.
- Incubate with an HRP-conjugated secondary antibody for 1 hour at room temperature.
- Wash the membrane 3 times for 5 minutes with TBST.
- Develop using a chemiluminescent substrate and image.
- To confirm equal loading, strip the membrane and re-probe with an antibody against the total substrate protein.
Protocol 2: Phosphopeptide Enrichment and TMT-based Quantitative Phosphoproteomics
This protocol provides an overview of a streamlined (SL-TMT) workflow for identifying and quantifying phosphorylation changes across multiple samples.
Materials:
- Cell pellets from treated/untreated conditions
- Urea lysis buffer (8 M Urea, 50 mM Tris-HCl, pH 8.0)
- Reduction and alkylation reagents (e.g., DTT and iodoacetamide)
- Lys-C and trypsin proteases
- Tandem Mass Tag (TMT) reagents
- Phosphopeptide enrichment resin (e.g., Fe³⁺-IMAC or TiO₂)
- StageTips for desalting
- LC-MS/MS system
Procedure:
- Cell Lysis and Digestion:
- Lyse cells in urea buffer. Reduce disulfide bonds with 5 mM DTT (30 min, RT) and alkylate with 15 mM iodoacetamide (30 min, RT in the dark).
- Perform methanol-chloroform precipitation to extract and purify proteins.
- Digest the proteins first with Lys-C (3 hours, RT) then dilute the urea concentration and digest with trypsin (overnight, 37°C).
TMT Labeling and Pooling:
- Label the resulting peptides from each condition with a different TMT reagent.
- Quench the reaction with hydroxylamine.
- Combine the TMT-labeled samples at a 1:1 ratio across all conditions.
Phosphopeptide Enrichment:
- Desalt the pooled peptide sample.
- Subject the sample to phosphopeptide enrichment using an IMAC or TiO₂ protocol.
- For IMAC: Incubate the peptide pool with Fe³⁺ or Ti⁴⁺-charged beads in a loading buffer (e.g., containing 0.1% TFA and 80% acetonitrile). Wash beads to remove non-specifically bound peptides. Elute phosphopeptides with an alkaline solution (e.g., pH 10-11) or a phosphate buffer.
LC-MS/MS Analysis and Data Processing:
- Acidify the eluted phosphopeptides and desalt using StageTips.
- Analyze by LC-MS/MS using a method capable of performing SPS-MS3 for accurate TMT quantification.
- Search the resulting spectra against a protein sequence database.
- Use bioinformatic tools to localize phosphorylation sites and quantify phosphorylation changes across conditions.
Visualization of Workflows
Diagram 1: Phosphoproteomic Workflow with TMT
Diagram 2: Peptide Library Screening Strategies
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents and Kits for Kinase Substrate Research
| Reagent/Kits | Function & Application | Example Use-Case |
|---|---|---|
| Phospho-Specific Antibodies | Immunodetection of specific phosphorylation sites in Western blot, ELISA, and ICC/IHC. | Validating phosphorylation of a putative substrate at a known residue (e.g., p53 Ser15) [9]. |
| Universal Kinase Activity Kit | Non-radioactive quantification of kinase activity by measuring ADP production. Applicable to any kinase. | Profiling inhibitor potency against a purified kinase using a generic substrate [9]. |
| Tandem Mass Tag (TMT) Reagents | Isobaric labels for multiplexed relative quantification of peptides and phosphopeptides from up to 16 samples in a single MS run. | Quantifying temporal changes in the phosphoproteome after kinase inhibitor treatment [57]. |
| Phosphopeptide Enrichment Kits (IMAC/MOAC) | Selective isolation of phosphopeptides from complex peptide digests prior to MS analysis. | Deepening coverage of the phosphoproteome by reducing sample complexity [57]. |
| Simple Western Capillary Systems | Automated, capillary-based Western blot analysis for highly sensitive and quantitative detection of phospho- and total-protein isoforms. | Precisely quantifying the activation loop phosphorylation of ERK1/2 with high sensitivity and reproducibility [61]. |
| Phospho-Specific ELISA Kits | Quantitative, microplate-based immunoassay for measuring specific phosphorylation events. | High-throughput screening of kinase inhibitor compounds in a cellular context [9]. |
| Peptide Library Platforms | Diverse collections of peptides for defining kinase specificity motifs. | Identifying the optimal substrate sequence for a novel or poorly characterized kinase [2] [59]. |
The strategic selection of substrate identification methods is critical for advancing kinase research. While traditional in vitro kinase assays remain the gold standard for validation, high-throughput peptide library and phosphoproteomic approaches have revolutionized our ability to discover novel substrates and define kinase specificity on a global scale. The integration of these methods, supported by robust enrichment techniques, sensitive detection reagents, and quantitative mass spectrometry, provides a comprehensive toolkit for researchers. This enables a deeper understanding of signaling networks in health and disease, accelerating the development of novel kinase-targeted therapies.
Assay Robustness: Troubleshooting Common Pitfalls and Optimization Strategies
In the realm of biochemical assay development for kinase activity and phosphorylation detection, fluorescence-based methodologies offer unparalleled sensitivity and real-time monitoring capabilities. However, the accuracy of these assays is frequently compromised by two predominant interference phenomena: fluorescence quenching and inner filter effects (IFE). These artifacts can significantly distort signal output, leading to inaccurate quantification of enzymatic activity and potentially flawed scientific conclusions. For researchers and drug development professionals working with kinase assays, understanding and mitigating these interference mechanisms is paramount for generating reliable, reproducible data, particularly when screening compound libraries where interferents may be prevalent.
Fluorescence quenching encompasses any process that decreases the fluorescence intensity of a fluorophore through intermolecular interactions, including excited-state reactions, energy transfer, ground-state complex formation, and collisional quenching [62]. In the specific context of kinase activity assays, quenching can occur when small molecules or compounds present in screening libraries directly interact with fluorophores, diminishing their emission intensity independent of the enzymatic reaction. This interference can manifest as false negatives or underestimated enzyme activity in high-throughput screening campaigns.
Inner filter effects represent a separate class of interference wherein the absorption of excitation or emission light by compounds in the assay solution reduces the detected fluorescence signal [63]. This phenomenon is particularly problematic in kinase assays that employ chromogenic ATP analogs or those conducted in complex biological matrices where absorbing species may be present. The IFE is distinct from quenching in that it does not involve direct interaction with the fluorophore's excited state but rather attenuates the light signals essential for fluorescence detection [64]. Both phenomena, if unaddressed, can severely compromise the linear relationship between fluorescence intensity and analyte concentration that forms the basis of robust quantitative assays.
Theoretical Foundations
Fluorescence Quenching Mechanisms
Fluorescence quenching mechanisms are broadly categorized into dynamic (collisional) and static quenching, each with distinct characteristics and implications for assay interference [62]. Dynamic quenching occurs when the excited-state fluorophore experiences contact with a quencher molecule, facilitating non-radiative return to the ground state through energy transfer [65]. This process requires diffusion and collision between fluorophore and quencher during the fluorescence lifetime. In contrast, static quenching involves the formation of a non-fluorescent complex between the fluorophore and quencher in the ground state, effectively reducing the population of excitable fluorophores [62] [65].
The Stern-Volmer equation describes the relationship between fluorescence intensity and quencher concentration for dynamic quenching:
[ \frac{F0}{F} = 1 + K{SV}[Q] = 1 + Kq \tau0 [Q] ]
Where (F0) and (F) represent fluorescence intensities in the absence and presence of quencher, respectively, (K{SV}) is the Stern-Volmer quenching constant, ([Q]) is the quencher concentration, (Kq) is the bimolecular quenching constant, and (\tau0) is the excited-state lifetime of the fluorophore in the absence of quencher [62].
Table 1: Key Characteristics of Quenching Mechanisms
| Parameter | Dynamic Quenching | Static Quenching |
|---|---|---|
| Temperature Effect | Increases with temperature | Decreases with temperature |
| Fluorescence Lifetime | Decreased ((\tau_0/\tau > 1)) | Unchanged ((\tau_0/\tau = 1)) |
| Stern-Volmer Plot | Linear | Linear (at low concentrations) |
| Bimolecular Quenching Constant (K_q) | Typically ≤ 2.0×10^10 L·mol⁻¹·s⁻¹ | Typically > 2.0×10^10 L·mol⁻¹·s⁻¹ |
Differentiation between these mechanisms is crucial for developing effective mitigation strategies. Dynamic quenching is confirmed through measurement of decreased fluorescence lifetime, while static quenching preserves the lifetime of the uncomplexed fluorophores [62]. For kinase assays, this distinction informs whether interference arises from transient interactions (potentially addressable by modifying assay kinetics) or stable complex formation (requiring compound structural modification or alternative detection chemistries).
Inner Filter Effects: Primary and Secondary
Inner filter effects are categorized as primary or secondary based on the light path affected. The primary inner filter effect (pIFE) occurs when compounds in the solution absorb the excitation light before it reaches the fluorophore, effectively reducing the number of photons available to excite the fluorophore [64]. This becomes significant when the absorbance of the solution at the excitation wavelength exceeds approximately 0.1, and is particularly problematic in assays employing high compound concentrations or those with inherently strong chromophores.
The secondary inner filter effect (sIFE) arises when emitted fluorescence is reabsorbed by compounds in the solution before reaching the detector [64]. This occurs when the absorption spectrum of an interfering compound overlaps with the emission spectrum of the fluorophore. The sIFE can lead to both signal attenuation and distortion of emission spectra, including peak shifts that complicate spectral interpretation.
The corrected fluorescence intensity ((F_{corr})) accounting for inner filter effects can be calculated using the formula:
[ \frac{F{corr}}{F{obs}} = 10^{\frac{d{ex}A{ex} + d{em}A{em}}{L}} ]
Where (F{obs}) is the measured fluorescence intensity, (A{ex}) and (A{em}) represent absorbance at excitation and emission wavelengths, respectively, (d{ex}) and (d{em}) are the excitation and emission path lengths, and (L) is the cuvette path length [64]. Advanced correction methods introduce an optimized parameter (n{opt}) that reflects solute-solvent system-specific self-absorption effects, providing more accurate compensation for sIFE [64].
Experimental Protocols for Identification and Mitigation
Protocol 1: Diagnosing Quenching Mechanisms in Kinase Assays
Purpose: To distinguish between dynamic and static quenching interference from compounds in kinase activity assays.
Materials:
- Fluorescent kinase substrate (e.g., coumarin-derivatized peptide)
- Test compounds (potential interferents)
- Kinase enzyme of interest
- ATP solution
- Fluorescence spectrometer with time-resolved capability
- Microplate reader or cuvette-based fluorimeter
Procedure:
- Prepare a solution containing the fluorescent substrate at the working concentration used in your kinase assay.
- Divide the solution into aliquots and add increasing concentrations of test compounds (typically 0.1-100 µM).
- Measure fluorescence intensity ((F)) for each sample using the same excitation/emission wavelengths as your kinase assay.
- Generate a Stern-Volmer plot by graphing (F0/F) versus quencher concentration [Q], where (F0) is fluorescence without quencher.
- For linear Stern-Volmer plots, calculate (K_{SV}) from the slope.
- Repeat measurements at different temperatures (e.g., 25°C and 37°C):
- If (K{SV}) increases with temperature → dynamic quenching
- If (K{SV}) decreases with temperature → static quenching [62]
- Confirm mechanism by measuring fluorescence lifetimes where equipment permits:
- Decreased lifetime indicates dynamic quenching
- Unchanged lifetime indicates static quenching [62]
Interpretation: Dynamic quenching suggests transient interactions potentially manageable by adjusting assay timing or temperature. Static quenching indicates stable complex formation, likely requiring compound structural modification or alternative detection strategies.
Protocol 2: Quantification and Correction of Inner Filter Effects
Purpose: To measure and correct for inner filter effects in compound-based kinase assays.
Materials:
- Fluorophore solution (at standard assay concentration)
- Test compounds
- UV-Vis spectrophotometer
- Fluorescence spectrometer
- Microplates or cuvettes compatible with both instruments
Procedure:
- Prepare compound solutions at the highest concentration used in screening.
- Measure absorbance spectra of each compound solution from 220-700 nm.
- Identify absorbance at your assay's excitation and emission wavelengths ((A{ex}) and (A{em})).
- Prepare fluorophore solutions with and without compounds at screening concentrations.
- Measure fluorescence intensity ((F_{obs})) for all samples.
- Calculate corrected fluorescence ((F{corr})) using the formula: [ F{corr} = F{obs} \times 10^{\frac{A{ex} + A{em}}{2}} ] For more accurate correction, use instrument-specific path lengths: [ F{corr} = F{obs} \times 10^{\frac{d{ex}A{ex} + d{em}A{em}}{L}} ] where (d{ex}), (d_{em}), and (L) are determined experimentally [64].
- For advanced correction of sIFE, determine the system-specific parameter (n_{opt}) through iterative fitting to expand the linear concentration range [64].
Troubleshooting: If corrected values still show concentration-dependent deviations, consider dye aggregation effects or the formation of exciplexes that may require more sophisticated correction approaches.
Protocol 3: Validation of Kinase Activity Assays Against Interference
Purpose: To confirm that measured kinase inhibition results from true enzymatic inhibition rather than interference phenomena.
Materials:
- Active kinase enzyme
- Inactive kinase (heat-inactivated or catalytic mutant)
- Fluorescent substrate
- ATP
- Putative inhibitor compounds
- Reference inhibitor (e.g., staurosporine)
- EDTA (for reaction quenching)
Procedure:
- Set up parallel reaction series with active and inactive kinase:
- Active kinase + substrate + ATP + compound
- Inactive kinase + substrate + ATP + compound
- Controls: no compound, reference inhibitor
- Incubate under standard assay conditions.
- Stop reactions with EDTA at various time points.
- Measure fluorescence intensity for all samples.
- Calculate apparent inhibition for both active and inactive kinase setups: [ \%\,Inhibition = \left(1 - \frac{F{compound} - F{background}}{F{control} - F{background}}\right) \times 100 ]
- Compare inhibition values between active and inactive kinase:
- True inhibition: Significant inhibition only with active kinase
- Interference: Similar "inhibition" with both active and inactive kinase
- For compounds showing interference, apply quenching and IFE corrections from Protocols 1 and 2.
Validation: True inhibitors should demonstrate concentration-dependent inhibition exclusively in active kinase samples, with minimal signal modulation in inactive kinase controls.
Research Reagent Solutions for Interference-Resistant Assays
Table 2: Essential Reagents for Interference-Mitigated Kinase Assays
| Reagent Category | Specific Examples | Function in Mitigation | Application Notes |
|---|---|---|---|
| Long-Lifetime Fluorophores | LanthaScreen Tb-labeled antibody [66], Ruthenium complexes | Enables time-resolved detection to eliminate short-lived background | Reduces compound autofluorescence interference |
| FRET-Compatible Dyes | Coumarin derivatives, Dark quenchers (e.g., IRDye QC-1) [65] | Provides built-in rationetric correction | Minimizes concentration-dependent artifacts |
| Reference Fluorophores | Fluorescein sodium, Rhodamine B [64] | Internal calibration standards | Corrects for path length variations and IFE |
| Quencher Dyes | Acid Green 27 [67], QSY series | Positive controls for quenching studies | Validates quenching detection protocols |
| Specialized Substrates | Coumarin-Aoc-GRTGRRFSYP-amide [67] | Optimized spectral properties | Reduces overlap with compound absorbance |
| Correction Reagents | Non-absorbing buffers, Spectral calibration standards [64] | Quantification of background effects | Enables mathematical correction of residual IFE |
Data Analysis and Correction Workflows
The following workflow diagram illustrates the systematic approach to addressing fluorescence interference in kinase activity assays:
Spectral Interference Diagnosis and Correction Workflow
For data processing following experimental measurements, implement this stepwise correction algorithm:
Primary IFE Correction: [ F{corr1} = F{obs} \times 10^{\frac{A{ex} + A{em}}{2}} ]
Quenching Assessment:
- Plot (F_0/F) vs. [Q] for Stern-Volmer analysis
- Determine (K_{SV}) and temperature dependence
Advanced sIFE Correction (for systems with significant emission reabsorption): [ F{corr2} = F{corr1} \times 10^{\frac{n{opt} \times A{em}}{2}} ] where (n_{opt}) is determined empirically for specific fluorophore-solvent systems [64].
Validation:
- Compare corrected dose-response curves with reference inhibitors
- Confirm expected pharmacology (e.g., ATP-competition patterns)
Table 3: Quantitative Parameters for Interference Assessment
| Parameter | Acceptance Threshold | Correction Required | Critical Value |
|---|---|---|---|
| Absorbance at λ_ex | < 0.05 | > 0.1 | > 0.3 |
| Absorbance at λ_em | < 0.02 | > 0.05 | > 0.2 |
| Stern-Volmer Constant (K_SV) | < 100 M⁻¹ | 100-1000 M⁻¹ | > 1000 M⁻¹ |
| Temperature Dependence (K_SV ratio) | 0.8-1.2 | 1.2-2.0 or 0.5-0.8 | > 2.0 or < 0.5 |
| Z'-factor with Interferents | > 0.5 | 0.3-0.5 | < 0.3 |
Case Study: PKA Activity Sensing with Quenching-Based Detection
A highly effective approach to mitigating interference involves designing assays that incorporate quenching mechanisms into the detection strategy itself. A representative example is the PKA activity sensor developed by Rantanen et al., which employs a quenching-based mechanism with an unprecedented 150-fold dynamic range [67]. This sensor consists of three key components: (1) a coumarin-derivatized peptide substrate (Cou-Aoc-GRTGRRFSYP-amide), (2) the negatively charged quencher dye Acid Green 27, and (3) the 14-3-3 phospho-binding domain.
The assay mechanism relies on electrostatic interactions between the positively charged peptide substrate and the negatively charged quencher, which efficiently suppress coumarin fluorescence in the unphosphorylated state. Upon PKA-catalyzed phosphorylation, the phosphorylated peptide associates with the 14-3-3 domain, displacing the quencher and restoring fluorescence [67]. This design provides inherent resistance to compound interference because the signal generation depends on specific displacement rather than absolute intensity measurements.
Key optimization parameters for this approach include:
- Electrostatic Optimization: The quenching efficiency demonstrated Arg residue dependence, with single Arg-to-Ala substitutions increasing KD values by approximately two orders of magnitude [67].
- Dynamic Range: The 152-fold fluorescence enhancement enables detection of PKA at concentrations as low as 160 pM, providing substantial signal window to distinguish true activity from interference [67].
- Biological Validation: The sensor successfully characterized the suborganelle distribution of PKA activity in bovine heart mitochondria (85% matrix, 6% intermembrane space, 9% outer membrane) [67], demonstrating utility in complex biological environments.
This case study illustrates how incorporating quenching into assay design, rather than merely correcting for it, can create robust systems resistant to compound interference while providing enhanced sensitivity for kinase activity quantification.
Implementation in High-Throughput Screening
For drug development professionals implementing kinase assays in high-throughput screening (HTS) environments, systematic interference mitigation is essential for maintaining data quality. The following dot language diagram illustrates an HTS-optimized workflow:
HTS Interference Correction Pipeline
Key implementation considerations for HTS include:
- Plate Design: Incorporate control wells for background subtraction, IFE calibration, and quenching assessment across each plate.
- Automated Absorbance Prediction: Implement computational tools to estimate compound absorbance based on structure to flag potential interferents before screening.
- Orthogonal Confirmation: Always follow up fluorescence-based screening with alternative detection technologies (e.g., AlphaScreen, TR-FRET, or mobility-shift assays) to confirm true inhibitors.
By integrating these interference-mitigation strategies directly into screening workflows, drug development teams can significantly reduce false positive rates and focus resources on compounds with genuine therapeutic potential.
Kinase activity assays are fundamental tools in biochemical research and drug discovery, enabling scientists to quantify enzyme function, study signaling pathways, and identify potential therapeutic inhibitors. These assays directly measure a kinase's ability to catalyze the transfer of a phosphate group from adenosine triphosphate (ATP) to specific protein substrates [9] [1]. The precise optimization of critical parameters—enzyme, substrate, and ATP concentrations—is paramount for developing robust, sensitive, and reproducible assays suitable for high-throughput screening (HTS) and lead optimization [68]. A well-optimized assay minimizes variability, enhances throughput, and ensures interpretable data, forming the cornerstone of reliable kinase research [68]. This protocol details the systematic optimization of these core parameters within the context of kinase activity and phosphorylation detection, providing researchers with a standardized framework for assay development.
Fundamental Principles of Kinase Assays
Kinase Function and Biochemical Reaction
Kinases are enzymes that catalyze the transfer of the gamma (γ)-phosphate group from ATP to serine, threonine, or tyrosine residues on specific protein substrates. This phosphorylation reaction is a crucial post-translational modification that regulates a vast array of cellular activities, including the cell cycle, differentiation, metabolism, and signal transduction [9]. The general biochemical reaction can be summarized as: Protein Substrate + ATP → Phosphorylated Protein + ADP
The activity of a kinase is therefore quantified by measuring the formation of the phosphorylated product or the concomitant generation of adenosine diphosphate (ADP) [68] [1]. Abnormal phosphorylation events are implicated in numerous disease states, particularly cancer, making kinases prominent drug targets [9] [1].
Key Assay Formats and Detection Methods
Multiple assay formats are employed to detect kinase activity, each with unique advantages and considerations. These can be broadly categorized into activity assays and binding assays [1].
Activity Assays: These directly measure the catalytic function of kinases.
- ADP Detection Assays (e.g., Transcreener): These are "universal" assays that detect the ADP produced in the kinase reaction using competitive immunodetection with fluorescent readouts (FI, FP, TR-FRET). They are applicable to a broad range of kinases and are amenable to HTS due to their homogeneous "mix-and-read" format [68].
- Phosphosubstrate Detection Assays: These detect the phosphorylated peptide or protein product, often using phospho-specific antibodies in formats like ELISA, Western Blot, or TR-FRET [9].
- Mobility Shift Assays (e.g., KiMSA): These separate phosphorylated from non-phosphorylated fluorescently-labeled substrates based on charge differences using electrophoresis. The Kinase Mobility Shift Assay (KiMSA) is a non-radioactive method that offers high sensitivity and quantitative precision [37].
- Coupled/Luminescence Assays (e.g., ADP-Glo): These couple ADP production to a luciferase reaction that generates luminescence [1].
Binding Assays: These assess the binding affinity of small molecules to the kinase, often to the ATP-binding site, and include techniques like Fluorescence Polarization (FP) and Surface Plasmon Resonance (SPR) [68] [1].
The selection of an appropriate assay format depends on the specific research question, desired throughput, sensitivity requirements, and available instrumentation.
Experimental Workflow for Kinase Assay Optimization
The following diagram illustrates the logical workflow for developing and optimizing a biochemical kinase assay.
The Scientist's Toolkit: Key Research Reagent Solutions
Successful kinase assay development relies on a suite of specialized reagents and tools. The table below details essential materials and their functions.
Table 1: Essential Reagents for Kinase Activity Assays
| Item | Function/Description | Example/Note |
|---|---|---|
| Recombinant Kinase | The enzyme of interest, catalyzing the phosphorylation reaction. | Purified, active form is critical. Concentration must be determined empirically [68]. |
| Peptide/Protein Substrate | The target molecule that accepts the phosphate group from ATP. | Synthetic peptides (e.g., Kemptide for PKA [37]) or full-length proteins. |
| Adenosine Triphosphate (ATP) | The phosphate group donor. Its concentration is a key kinetic parameter. | A critical parameter to optimize; used at or below Km for inhibitor studies [68] [1]. |
| Detection Kit/Reagents | For quantifying reaction output (ADP or phosphorylated product). | Transcreener (ADP detection) [68]; Antibodies for phospho-ELISA/Western blot [9]. |
| Buffer Components | Maintain optimal pH, ionic strength, and provide essential cofactors. | HEPES or Tris, Mg2+ or Mn2+, DTT, BSA. Must be optimized for each kinase [68] [69]. |
| Positive Control Inhibitor | A known inhibitor to validate assay function and sensitivity. | e.g., Staurosporine or a specific, well-characterized inhibitor for the target kinase. |
| Fluorescent-Labeled Tracer | Binds to the detection antibody, generating a signal that is displaced by ADP. | Used in immuno-detection assays like Transcreener [68]. |
| Detection Antibody | Binds specifically to the analyte (e.g., ADP) or phosphorylated product. | High specificity and affinity are required for a robust signal [68] [9]. |
Core Optimization Parameters and Experimental Protocols
Systematic Optimization Using Design of Experiments (DoE)
Traditional one-factor-at-a-time (OFAT) optimization can be time-consuming and may miss interactions between factors. The Design of Experiments (DoE) approach systematically varies multiple parameters simultaneously, enabling the identification of optimal conditions and factor interactions in a more efficient manner. One study demonstrated that DoE could reduce the assay optimization process from over 12 weeks to less than 3 days [69]. This protocol will utilize a DoE framework for optimizing the core parameters.
Optimizing Enzyme Concentration
The goal is to determine the minimum enzyme concentration that yields a robust, linear signal over time, maximizing the signal-to-background (S/B) ratio while conserving precious enzyme reagents.
Protocol 4.2.1: Enzyme Titration for an ADP Detection Assay
- Reagent Preparation: Prepare a master mix containing assay buffer, ATP (at a fixed concentration near its apparent Km, e.g., 10 µM), substrate (at a fixed concentration, e.g., 1-5x Km), and MgCl₂. Dispense equal volumes into a 96-well or 384-well assay plate.
- Enzyme Dilution Series: Prepare a serial dilution of the kinase in assay buffer, covering a broad range (e.g., 0.1 nM to 100 nM). Include a "no-enzyme" control (background signal).
- Reaction Initiation: Initiate the reactions by adding the enzyme dilutions to the plate. Seal the plate and incubate at room temperature or 30°C for a predetermined time (e.g., 60 minutes).
- Reaction Termination and Detection: Stop the reaction by adding the detection mix (containing ADP-specific antibody and fluorescent tracer). Incubate according to kit specifications (e.g., 30-60 minutes).
- Signal Measurement: Read the plate using a compatible plate reader (e.g., for FI, FP, or TR-FRET).
- Data Analysis: Plot the signal (or % Inhibition if using a control) against the log of enzyme concentration. The optimal enzyme concentration is typically in the linear range of the curve, providing a high S/B ratio (often >3:1) without depleting the substrate excessively.
Table 2: Example Data from an Enzyme Titration Experiment
| Enzyme Concentration (nM) | Signal (RFU) | Background (RFU) | Signal-to-Background (S/B) |
|---|---|---|---|
| 0.0 | 15,000 | 15,000 | 1.0 |
| 0.1 | 18,500 | 15,000 | 1.2 |
| 0.5 | 28,000 | 15,000 | 1.9 |
| 1.0 | 41,000 | 15,000 | 2.7 |
| 2.5 | 55,000 | 15,000 | 3.7 |
| 5.0 | 62,000 | 15,000 | 4.1 |
| 10.0 | 65,000 | 15,000 | 4.3 |
In this example, 2.5 nM enzyme is selected for further assay development as it provides a strong S/B > 3 while using a conservative amount of enzyme.
Optimizing Substrate and ATP Concentrations
The Michaelis-Menten constant (Km) defines the substrate concentration at which the reaction velocity is half of Vmax. Determining the apparent Km for both the peptide substrate and ATP is essential for setting up sensitive and physiologically relevant assay conditions, especially for inhibitor studies.
Protocol 4.3.1: Determining Apparent Km for Substrate and ATP This protocol uses a DoE approach by titrating both substrate and ATP in a matrix format.
- Setup: Using the optimized enzyme concentration from Protocol 4.2.1, prepare a two-dimensional titration. For the substrate, test a range from 0 to 50 µM (e.g., 0, 0.5, 1, 5, 10, 25, 50 µM). For ATP, test a range from 0 to 100 µM (e.g., 0, 1, 5, 10, 25, 50, 100 µM).
- Reaction and Detection: Combine reagents to initiate the kinase reaction as before. Stop the reaction and detect the signal using the chosen method.
- Data Analysis: For each fixed ATP concentration, plot the initial velocity (V0) against the substrate concentration and fit the data to the Michaelis-Menten equation to determine the apparent Km(substrate). Repeat the analysis for ATP Km by holding substrate concentration fixed. The optimal assay condition for screening is often run with substrate and ATP concentrations at or below their apparent Km values to maximize sensitivity to competitive inhibitors [68] [70].
Table 3: Example of Apparent Km Determination for a Peptide Substrate
| [Substrate] (µM) | Velocity V0 (RFU/min) | Notes |
|---|---|---|
| 0 | 0 | Background, no reaction |
| 0.5 | 850 | |
| 1.0 | 1,550 | |
| 2.5 | 3,000 | |
| 5.0 | 4,500 | |
| 10.0 | 5,800 | |
| 25.0 | 6,900 | Near Vmax |
| 50.0 | 7,100 | Vmax |
Non-linear regression analysis of this data yields an apparent Km of ~4.5 µM. For a screening assay, a substrate concentration of 5 µM (slightly above Km) would be suitable.
Detailed Protocol: Kinase Mobility Shift Assay (KiMSA)
The KiMSA is a powerful, non-radioactive alternative that directly visualizes substrate phosphorylation via a change in electrophoretic mobility [37].
Protocol 4.4.1: KiMSA for PKA Activity Measurement
- Cell Lysis and Extract Preparation: Prepare sperm cells or other biological material. Lyse cells in an appropriate buffer containing protease and phosphatase inhibitors. Centrifuge to clear debris and collect the supernatant (total extract) [37].
- Kinase Reaction Setup:
- Component: Final Concentration/Amount
- Total Cell Extract: 10-20 µg protein
- Kinase Buffer (HEPES, MgCl₂): 1X
- Kemptide-FITC: 5 µM
- ATP: 100 µM
- Water: to final volume
- Incubate for 25 minutes at 37°C in the dark [37].
- Reaction Termination: Stop the reaction by placing tubes on ice. Add Tween-20 to a final concentration of 0.01% and incubate at 100°C for 1 minute.
- Electrophoresis: Load the reactions onto an agarose gel. Run the electrophoresis under appropriate conditions to separate phosphorylated and non-phosphorylated Kemptide-FITC.
- Visualization and Quantification: Image the gel using a fluorescence scanner. Quantify the band intensities using densitometry software. Calculate PKA activity as the percentage of phosphorylated Kemptide-FITC or as International Units (IU) of normalized activity [37].
The workflow for this specific protocol is outlined below.
Validation and Data Analysis
Assessing Assay Quality and Robustness
Once optimal conditions are established, the assay must be validated for performance and robustness. The Z'-factor is a standard statistical parameter used for this purpose in HTS. It assesses the quality of the assay by accounting for the dynamic range and data variation of both positive and negative controls [68]. Formula: Z' = 1 - [3*(σp + σn) / |μp - μn|] Where σp and σn are the standard deviations of the positive (no inhibition, high signal) and negative (full inhibition, low signal) controls, and μp and μn are their respective means. A Z'-factor > 0.5 indicates an excellent assay robust enough for HTS [68].
Advanced Estimation of Inhibition Constants
Traditional methods for estimating inhibition constants (Kic, Kiu) require extensive experiments at multiple substrate and inhibitor concentrations. A recent advancement, the 50-BOA (IC50-Based Optimal Approach), demonstrates that precise estimation is possible with a single inhibitor concentration greater than the IC50, dramatically reducing the number of required experiments by over 75% [70]. This method incorporates the relationship between IC50 and the inhibition constants into the fitting process of the velocity equation, ensuring accuracy and precision while improving efficiency [70].
The rigorous optimization of enzyme, substrate, and ATP concentrations is a critical, non-negotiable step in developing reliable and predictive kinase activity assays. By following the systematic protocols outlined in this application note—employing strategic titrations, leveraging universal assay platforms where appropriate, and utilizing advanced statistical methods like DoE and 50-BOA—researchers can accelerate their assay development process. This approach ensures the generation of high-quality, reproducible data that is essential for driving fundamental kinase research and advancing drug discovery pipelines. A well-optimized assay is not merely a technical exercise; it is the foundation upon which sound scientific conclusions and therapeutic breakthroughs are built.
Dimethyl sulfoxide (DMSO) is an indispensable solvent in kinase research, particularly for dissolving hydrophobic small-molecule inhibitors during biochemical assays. Its amphipathic nature allows it to solubilize both polar and nonpolar compounds, making it invaluable for pharmaceutical applications. However, DMSO is not physiologically inert and can significantly influence experimental outcomes by affecting protein conformation, enzymatic kinetics, and signal detection. Understanding and managing DMSO tolerance is therefore critical for generating reliable, reproducible data in kinase activity and phosphorylation studies. This application note provides a structured framework for quantifying DMSO effects, establishing compatible concentration thresholds, and implementing optimized experimental protocols to control for solvent-related artifacts in kinase research.
Quantitative DMSO Tolerance in Biological and Biochemical Systems
DMSO tolerance varies significantly across different experimental systems. The following tables summarize established concentration thresholds from recent studies.
Table 1: DMSO Tolerance in Model Organisms and Cellular Systems
| System | Safe Concentration | Observed Effects | Critical Endpoints | Citation |
|---|---|---|---|---|
| Zebrafish Embryos | ≤1% (v/v) | Morphological & physiological alterations (1-4%); Lethality (≥5%) | Pericardial edema, tail curvature, reduced heart rate, somite defects | [71] |
| Fungal Pathogens (F. graminearum) | <0.5% (v/v) | Inhibited mycelial growth & conidial germination | Reduced pathogenicity, altered pigmentation, suppressed reproduction | [72] |
| Enzymatic Bioluminescence (BLuc–Red) | 4-6% (v/v) | Partial inhibition at higher concentrations | Luminescence suppression, altered enzyme flexibility | [73] |
| Human Nerve Growth Factor (hNGF) | ≤0.8% (v/v) | No significant conformational changes; low-affinity binding | Preserved receptor-binding dynamics | [74] |
Table 2: DMSO Effects in Kinase Assays and Protein Studies
| Assay/Protein Context | Typical Working Concentration | Key Considerations | Impact on Kinase Activity | Citation |
|---|---|---|---|---|
| HotSpot Kinase Assay | 1% (v/v) | Standard component of base reaction buffer | Minimal impact at recommended level | [75] |
| General Kinase Assay Buffers | 0.1% - 1% (v/v) | Must be optimized for each kinase | Can be inhibitory or alter kinetics at high concentrations | [1] |
| Protein-Ligand Binding (hNGF) | ≤0.8% (v/v) | Binds with low affinity without disrupting structure | Potential to compete with or influence small-molecule binding | [74] |
| Recombinant Enzymes | Varies (1-10%) | Purity and source influence sensitivity | Solvent-induced conformational changes can modulate activity | [73] [74] |
Experimental Protocols for DMSO Tolerance Assessment
Protocol for Determining Maximum Tolerated DMSO in Kinase Assays
This protocol outlines a systematic approach to establish the maximum DMSO concentration that does not significantly inhibit kinase activity.
I. Materials
- Purified kinase of interest
- Kinase substrate (e.g., peptide or protein)
- ATP solution
- Kinase reaction buffer (e.g., 20 mM HEPES pH 7.5, 10 mM MgCl₂, 1 mM EGTA, 0.01% Brij35, 0.02 mg/ml BSA, 0.1 mM Na₃VO₄, 2 mM DTT) [75]
- DMSO (high-grade, spectrophotometric grade)
- Detection reagents (e.g., ADP-Glo kit or phosphospecific antibodies)
II. Procedure
- Preparation of DMSO Dilution Series: Create a dilution series of DMSO in the kinase reaction buffer. A typical series includes 0%, 0.5%, 1%, 2%, 4%, and 6% (v/v) DMSO.
- Reaction Setup:
- In a 96-well plate, combine the following components in sequence:
- 20 µL of kinase reaction buffer containing the appropriate DMSO concentration
- 5 µL of substrate solution
- 5 µL of kinase solution
- Pre-incubate the mixture for 20 minutes at room temperature.
- Initiate the reaction by adding 5 µL of ATP solution.
- Incubate for the predetermined optimal time (e.g., 2 hours at room temperature) [75].
- In a 96-well plate, combine the following components in sequence:
- Reaction Termination and Detection:
- Stop the reaction according to the detection method used.
- For luminescence-based detection (e.g., ADP-Glo), add an equal volume of detection reagent and incubate before measuring luminescence.
- Data Analysis:
- Normalize kinase activity at each DMSO concentration to the 0% DMSO control.
- Plot normalized activity (%) versus DMSO concentration (%).
- The maximum tolerated DMSO concentration is defined as the highest concentration where activity remains ≥90% of the control.
Protocol for Assessing DMSO-Protein Interactions via Differential Scanning Fluorimetry (DSF)
DSF is a powerful method to monitor DMSO-induced effects on protein thermal stability, which can correlate with functional changes.
I. Materials
- Purified protein (kinase or relevant protein, e.g., hNGF at ~1-5 mg/mL) [74]
- DSF-compatible fluorescent dye (e.g., SYPRO Orange)
- Real-time PCR instrument
- DMSO (high-grade)
- Appropriate protein storage buffer
II. Procedure
- Sample Preparation:
- Prepare a 5X concentrated stock solution of SYPRO Orange dye in buffer.
- In a PCR plate, mix:
- 18 µL of protein solution (with varying DMSO concentrations)
- 2 µL of 5X SYPRO Orange dye
- Final DMSO concentrations should span a range (e.g., 0%, 0.5%, 1%, 2%, 5%).
- Thermal Denaturation:
- Seal the plate and centrifuge briefly.
- Run the thermal ramp program on the real-time PCR instrument (e.g., from 25°C to 95°C with a 1°C/min increment).
- Data Analysis:
- Determine the melting temperature (Tm) for each sample from the inflection point of the fluorescence curve.
- Plot Tm versus DMSO concentration. A significant shift in Tm (>1°C) indicates a stabilizing or destabilizing interaction between DMSO and the protein [74].
Experimental Workflow and Decision Pathway
The following diagrams outline the key experimental workflows for assessing DMSO compatibility.
DMSO Tolerance Workflow
Kinase Assay Optimization
Research Reagent Solutions
The following table details essential reagents and their functions for DMSO tolerance studies in kinase research.
Table 3: Key Research Reagents for DMSO Tolerance and Kinase Studies
| Reagent | Function/Application | Specific Example/Note | Citation |
|---|---|---|---|
| DMSO (High Purity) | Universal solvent for hydrophobic compounds | Use spectrophotometric grade; store under anhydrous conditions | [75] |
| Kinase Reaction Buffer | Maintains optimal pH and ionic strength | Typically contains HEPES, Mg²⁺, DTT, BSA, and Brij-35 | [75] |
| ADP-Glo Kit | Luminescent detection of kinase activity | Measures ADP formation; compatible with up to 1-2% DMSO | [1] |
| SYPRO Orange Dye | Fluorescent probe for protein denaturation | Used in DSF to measure thermal stability shifts | [74] |
| P81 Filter Membranes | Radiometric phosphorylation assay | Binds phosphorylated peptides; used in HotSpot assay | [75] |
| Positional Scanning Peptide Library | Profiling kinase substrate specificity | Commercial libraries available (e.g., Anaspec) | [76] |
| Recombinant Kinases | Catalytic component of kinase assays | Source and purity critically affect DMSO sensitivity | [1] |
Application in Kinase Research: Best Practices
Integrating DMSO tolerance data into kinase research requires careful experimental design and validation. The following guidelines ensure reliable results:
- Standardize DMSO Concentration: Maintain a consistent, low DMSO concentration (typically ≤1%) across all samples in an experiment, including controls, to avoid concentration-dependent artifacts [75] [1]. Use acoustic dispensing (e.g., Echo550) for nanoliter-volume transfers of compound-DMSO solutions to ensure accuracy and minimize final DMSO percentages [75].
- Validate with Orthogonal Assays: Confirm key findings, especially inhibitor IC₅₀ values, using a second kinase assay format based on a different detection principle (e.g., radiometric filter binding and TR-FRET) to rule out DMSO-interference with the detection system [1].
- Include Comprehensive Controls: Design control experiments that account for DMSO's potential effects. These include no-enzyme controls (background), no-substrate controls (non-specific phosphorylation), and vehicle controls (DMSO-only).
- Pre-test New Systems: Before initiating large-scale compound screening, always perform a DMSO tolerance curve with the specific kinase and assay format to be used, as sensitivity can vary significantly between kinases and detection methods [1].
- Monitor Protein Stability: For sensitive kinases or long-term assays, utilize DSF to ensure your working DMSO concentration does not destabilize the kinase structure, which could lead to loss of activity over time [74].
Mitigating Non-Specific Inhibition and Chelation Artifacts
Non-specific inhibition and metal chelation represent significant sources of artifactual results in kinase activity assays, potentially compromising data quality and leading to false conclusions in drug discovery pipelines. These artifacts arise when test compounds interfere with assay detection technology or disrupt biological systems through mechanisms independent of target kinase engagement [1] [77]. Within biochemical kinase assays, chelators can inadvertently sequester essential metal cofactors, such as Mg²⁺ and Mn²⁺, which are critical for ATP binding and catalytic transfer of phosphate groups to protein substrates [78]. The prevalence of these artifacts necessitates robust detection and mitigation strategies to ensure the identification of genuine kinase inhibitors and the accurate characterization of compound mechanism of action.
Understanding the Artifacts
Mechanisms of Non-Specific Inhibition
Non-specific inhibition in kinase assays manifests through several distinct mechanisms that can produce false positives or negatives. Compound interference can be broadly categorized into technology-related and biology-related artifacts [77].
- Technology-Related Interference: This includes compound autofluorescence, which can produce false positive signals in fluorescence-based detection systems, and fluorescence quenching, which can mask genuine signals and create false negatives. Additionally, colored or pigmented compounds can interfere with light transmission and reflection in spectrophotometric and imaging-based readouts [77].
- Biology-Related Interference: This encompasses non-specific chemical reactivity, colloidal aggregation, redox cycling, and denaturation mediated by surfactants or detergents. These mechanisms can lead to apparent inhibition without direct interaction with the kinase's active site [77]. Cytotoxic compounds or those that dramatically alter cellular morphology can also produce artifactual phenotypes in cell-based kinase assays [77].
Chelation Artifacts in Kinase Systems
Metal chelation artifacts occur when compounds sequester essential divalent cations required for kinase function. The kinase catalytic domain utilizes Mg²⁺ or Mn²⁺ ions to facilitate the transfer of the gamma-phosphate from ATP to substrate proteins [78]. Chelation can inhibit kinase activity non-specifically by depriving the enzyme of these essential cofactors. The resulting metal-chelator complex may itself possess biological activity, further complicating data interpretation [78]. The table below summarizes common metal chelators and their impacts on kinase activity assays.
Table 1: Common Chelators and Their Interference in Kinase Assays
| Chelator/Compound Type | Metals Targeted | Impact on Kinase Activity | Detection Challenges |
|---|---|---|---|
| EDTA / EGTA | Ca²⁺, Mg²⁺, Mn²⁺ | Broad-spectrum inhibition by depleting essential cofactors | Easy to identify due to known strong chelating property |
| Compounds with catechol, hydroxamate, or thiosemicarbazone motifs | Fe²⁺/³⁺, Cu²⁺, Zn²⁺ | Can inhibit metal-dependent kinases; may generate cytotoxic ROS | More subtle; requires specific counter-screens [78] |
| Redox-active metal chelates | Fe, Cu | Can generate reactive oxygen species (ROS), leading to oxidative damage and false cellular phenotypes | Masquerades as specific inhibition in cellular assays [78] |
| Colloidal aggregates | N/A | Non-specific sequestration of proteins, mimicking inhibition | Not true chelation, but produces similar artifactual inhibition [77] |
Detection and Mitigation Strategies
A multi-faceted approach is essential to identify and mitigate non-specific artifacts. The following workflow and protocols provide a systematic framework for researchers.
Experimental Protocol: Metal Add-back Assay for Chelation Detection
This protocol tests whether apparent inhibition is reversed by supplementing with excess metal ions, indicating a chelation artifact [77] [78].
Materials:
- Purified kinase protein
- Test compound(s)
- Kinase substrate and ATP
- Assay buffer (e.g., HEPES or Tris-based)
- Metal stock solutions: 1 M MgCl₂, 1 M MnCl₂, 100 mM CaCl₂
- DMSO (for compound solubilization)
- Detection reagents (e.g., ADP-Glo, fluorescent antibody)
Procedure:
- Prepare Compound Dilutions: Serially dilute the test compound in DMSO. Include a control well with DMSO only.
- Set Up Reaction Mixtures: In a 96-well assay plate, add:
- Assay buffer
- Kinase and substrate
- Condition A: 1-5 mM MgCl₂ (standard concentration)
- Condition B: 1-5 mM MgCl₂ + 10 mM MgCl₂ (high concentration)
- Condition C: 1-5 mM MgCl₂ + 1-2 mM MnCl₂
- Initiate Reaction: Add ATP to start the kinase reaction. The final ATP concentration should reflect the Km(ATP) for the kinase.
- Incubate: Incubate at 30°C for an appropriate time (e.g., 30-60 minutes).
- Detect and Analyze: Stop the reaction and quantify phosphorylation. Plot % kinase activity vs. compound concentration for each condition.
Interpretation: A significant rightward shift (reduced potency) of the dose-response curve in Conditions B or C suggests the compound's inhibition is mediated by chelation of metal ions.
Experimental Protocol: Orthogonal Assay for Non-Specific Interference
This protocol uses a different detection technology to identify compounds that interfere with the primary assay's readout system [1] [77].
Materials:
- Primary Assay Kit (e.g., Fluorescence Polarization, FP)
- Orthogonal Assay Kit (e.g., Luminescence-based like ADP-Glo, or Mobility Shift)
- Test compound(s)
- Source of kinase activity (purified kinase or cell lysate)
Procedure:
- Run Primary Assay: Perform the kinase assay with test compounds according to the standard protocol (e.g., FP assay).
- Run Orthogonal Assay: Using the same concentration of kinase, test compounds, and ATP, perform the kinase assay with the orthogonal technology.
- Control for Artifacts: Include control wells for autofluorescence/quenching in the FP assay and for luciferase inhibition in the ADP-Glo assay.
- Quantify and Compare: Calculate % inhibition for each compound in both assays.
Interpretation: A compound that shows strong inhibition in the primary assay but no activity in the orthogonal assay is likely a technology-specific interferent. Correlation of IC₅₀ values between the two platforms increases confidence in the hit.
Table 2: Key Research Reagent Solutions for Artifact Mitigation
| Reagent / Tool | Function | Application Context |
|---|---|---|
| ADP-Glo Kinase Assay | Luminescent detection of ADP formation; insensitive to optical interferents | Orthogonal assay for fluorescence-based primary screens; confirms catalytic inhibition [1] |
| TR-FRET/HTRF Kinase Assays | Time-resolved FRET detection with low background; reduces short-lifetime fluorescence interference | Primary or orthogonal assay; useful for screening compound libraries [1] |
| Mobility Shift Assays | Charge-based separation of phosphorylated/non-phosphorylated peptide; non-optical | Orthogonal assay to rule out fluorescence-based artifacts [1] |
| Prochelator Molecules | Chelators activated by disease-specific triggers (e.g., H₂O₂) | Research tool to study metal biology without causing systemic chelation artifacts [78] |
| Reference Interference Compounds | Known aggregators, fluorescent compounds, or chelators (e.g., rifampicin, rotenone) | Positive controls for interference counter-screens during assay development and validation [77] |
Data Analysis and Interpretation
Robust data analysis is critical for flagging potential artifacts. Statistical analysis of control well data and compound well readouts can identify outliers indicative of interference [77]. For example, compounds that cause dramatic changes in nuclear count or nuclear stain intensity in high-content screening are likely cytotoxic, while those producing signal intensities far outside the normal distribution may be fluorescent or quenching [77].
Table 3: Summary of Artifact Detection Methods and Data Interpretation
| Artifact Type | Key Detection Methods | Interpretation of Positive Result | Recommended Action |
|---|---|---|---|
| Metal Chelation | Metal add-back assay; correlation across metal-dependent and independent assays | Loss of potency with excess Mg²⁺/Mn²⁺ | Exclude or redesign compound to remove chelating motifs |
| Compound Autofluorescence | Compare signals in orthogonal assays (e.g., luminescence vs fluorescence); measure compound alone in assay buffer | High signal in fluorescent assay without kinase; no activity in luminescent assay | Flag as interferent; use orthogonal platform for validation |
| Fluorescence Quenching | Compare signals in orthogonal assays; measure signal loss from control wells | Low signal in fluorescent assay; confirmed activity in luminescent assay | Flag as interferent; use orthogonal platform for validation |
| Cellular Toxicity / Morphology Change | Statistical analysis of cell count and morphology parameters in HCS | Significant reduction in cell count or extreme morphological changes | Attribute phenotype to cytotoxicity, not specific kinase inhibition |
| Colloidal Aggregation | Detergent addition (e.g., Triton X-100, CHAPS); dynamic light scattering | Loss of inhibition in presence of 0.01% Triton X-100 | Exclude or redesign compound to improve solubility |
Vigilance against non-specific inhibition and chelation artifacts is a cornerstone of robust kinase research and drug discovery. By integrating careful assay design, systematic counter-screening, and intelligent data analysis, researchers can effectively triage artifactual compounds. The application of orthogonal assay formats and metal add-back experiments, as detailed in these protocols, provides a reliable path for confirming the specificity of kinase inhibitors. Adopting this rigorous, multi-pronged approach ensures that resources are focused on chemically tractable and biologically relevant lead compounds, ultimately accelerating the development of targeted kinase therapies.
Kinases represent one of the largest and most important families of drug targets, with approximately one-third of all drug development efforts focused on their inhibition [79]. However, researchers face significant challenges when working with kinases that require autophosphorylation for activation or those classified as "orphan" kinases with unknown substrates and regulatory mechanisms. These challenges are particularly pronounced in neglected tropical disease research where kinomes of pathogens like Plasmodium falciparum and Trypanosoma brucei contain numerous orphan kinases displaying no homology to known human kinases [80]. Establishing robust biochemical assays for such kinases is essential for targeted drug discovery and understanding their physiological functions.
Orphan kinases—those without clear orthologues in well-characterized kinomes—comprise substantial portions of pathogenic organism kinomes. For example, Trypanosoma brucei has over 30 orphan kinases, while Cryptosporidium parvum has 25% of its kinome classified as having no known orthologues outside of Cryptosporidium [80]. Similarly, in plants, many receptor-like kinases (RLKs) remain functionally uncharacterized with unknown substrates [81]. This knowledge gap severely hampers drug discovery efforts against these promising targets.
Table 1: Prevalence of Orphan Kinases in Various Organisms
| Organism | Kinome Size | Orphan Kinases | Reference |
|---|---|---|---|
| Trypanosoma brucei | 156-179 | >30 | [80] |
| Cryptosporidium parvum | ~70 | 25% of kinome | [80] |
| Entamoeba histolytica | >300 | 38 classified as 'other' | [80] |
| Plants (Arabidopsis) | >600 | Many RLKs uncharacterized | [81] |
Autophosphorylation Mechanisms and Regulatory Roles
Biological Significance of Autophosphorylation
Autophosphorylation—the process whereby a kinase catalyzes phosphotransfer to residues within its own structure—serves as a critical regulatory mechanism for many kinases. This self-modification typically occurs on the activation loop, either in trans (between kinase molecules) or cis (within the same molecule) fashion, often inducing conformational changes that enhance catalytic activity toward exogenous substrates [80]. Recent research has revealed unexpected complexity in autophosphorylation mechanisms, including dual-specificity autophosphorylation that expands regulatory possibilities beyond traditional serine/threonine or tyrosine phosphorylation.
The functional consequences of autophosphorylation are diverse and can significantly impact substrate specificity. For instance, Casein Kinase 1 (CK1) autophosphorylates a conserved threonine residue (T220 in human CK1δ) located at the N-terminus of helix αG, proximal to the substrate binding cleft [82]. This modification induces structural plasticity that alters the conformation of the substrate binding cleft, thereby affecting substrate specificity rather than uniformly enhancing or inhibiting activity toward all substrates.
Case Studies of Autophosphorylation
IKK2 Dual-Specificity Autophosphorylation: The inhibitor of nuclear factor kappa-B kinase subunit beta (IKK2/IKKβ), traditionally classified as a serine/threonine kinase, exhibits unexpected dual-specificity autophosphorylation at tyrosine residues [23]. Specifically, autophosphorylation at Y169 (located at the DFG+1 position) is essential for phosphorylating S32 within its IκBα substrate. Mutation of Y169 to phenylalanine severely compromises IκBα phosphorylation, demonstrating that tyrosine autophosphorylation enables substrate phosphorylation through a potential phospho-relay mechanism [23].
Calcium-Dependent Protein Kinases: CDPK3 from Toxoplasma gondii requires preincubation with ATP and CaCl₂ to auto-activate prior to activity measurements with exogenous substrates [80]. This autophosphorylation-dependent activation is sensitive to inhibitors like purfalcamine (IC₅₀ = 800 nM), highlighting the druggability of this regulatory mechanism.
Table 2: Characterized Autophosphorylation Events and Their Functional Consequences
| Kinase | Autophosphorylation Site | Functional Consequence | Reference |
|---|---|---|---|
| CK1δ | T220 | Alters substrate binding cleft conformation; modulates specificity | [82] |
| IKK2 | Y169 | Enables phosphorylation of IκBα at S32; essential for fidelity | [23] |
| C2PK (E. histolytica) | S428 | Regulates catalytic activity; inhibited by staurosporine (IC₅₀ 150 nM) | [80] |
| MAPK1 (T. gondii) | Multiple sites | Autokinase activity; inhibited by SB505124 (IC₅₀ 125 nM) | [80] |
Diagram 1: Autophosphorylation Activation Mechanism (65 characters)
Experimental Strategies for Orphan Kinases
Substrate Identification Approaches
Orphan kinases present a formidable challenge because their native physiological substrates are unknown. Fortunately, most kinases exhibit some degree of promiscuity and will catalyze phosphotransfer from ATP to alternative substrates, enabling the development of functional assays [80]. Several complementary approaches have proven successful for identifying substrates for orphan kinases:
Generic Substrate Screening: Initial screening with generic substrates like casein, myelin basic protein (MBP), histones, or synthetic peptides (kemptide, Syntide-2, Crosstide) can identify baseline catalytic activity [80]. Dephosphorylated casein is particularly useful for kinases with homology to casein kinases. This approach requires minimal prior knowledge of kinase specificity and provides a starting point for assay development.
Kinase-Client (KiC) Assay: This innovative approach exposes purified kinases to synthetic peptide libraries and detects phosphorylated peptides using liquid chromatography-tandem mass spectrometry (LC-MS/MS) [81]. The KiC assay assumes that amino acid sequences surrounding phosphorylation sites determine substrate specificity and provides superior signal-to-noise ratios compared to other large-scale methods. Recent advances incorporate machine learning predictions (e.g., MUsite) and hidden Markov models (HMMER) to design more diverse and informative peptide libraries [81].
Kinase Inhibition Profiling (KiPIK): This method exploits the selectivity profiles of kinase inhibitors as fingerprints to identify kinases responsible for specific phosphorylation events [83]. Cell extracts containing kinase activity are challenged with a panel of well-characterized inhibitors, and the resulting inhibition pattern is compared to known inhibition profiles of recombinant kinases. This approach successfully identified Aurora B as the kinase responsible for histone H3S28 phosphorylation in mitosis [83].
Computational and Structural Approaches
Advances in computational methods have significantly enhanced our ability to characterize orphan kinases. Deep learning models like PhoSiteformer and CSPred combine protein sequence embeddings with phosphorylation mass spectrometry data to systematically identify critical phosphorylation sites that enhance kinase activity [84]. This approach recently identified 77 critical phosphosites on 58 human kinases, expanding our understanding of kinase regulation [84].
For microbial orphan histidine kinases, evolutionary analyses combining structural, domain, sequence, and phylogenetic examinations can reveal origins and functional capabilities [85]. These analyses suggest that orphan histidine kinases and response regulators evolve through gene duplication, divergence, and domain shuffling processes, providing insights for functional prediction.
Diagram 2: Orphan Kinase Characterization Workflow (56 characters)
Detailed Experimental Protocols
Autophosphorylation-Dependent Kinase Activation Assay
Purpose: To measure and characterize autophosphorylation requirements for kinase activation and substrate phosphorylation.
Materials:
- Purified recombinant kinase (wild-type and phosphorylation site mutants)
- Kinase assay buffer (e.g., 25 mM Tris-HCl pH 7.5, 10 mM MgCl₂, 1 mM DTT)
- ATP solution (prepared fresh, varying concentrations)
- Calcium chloride (for CDPKs) or other activating compounds
- Lambda protein phosphatase (for dephosphorylation controls)
- Substrate (generic or putative specific substrate)
- [γ-³²P]ATP or ADP for phospho-relay detection [23]
- Stop solution (e.g., SDS-PAGE loading buffer)
Procedure:
- Kinase Dephosphorylation (Optional): Treat purified kinase with lambda phosphatase (400 units/µg kinase) for 30 minutes at 30°C to remove pre-existing phosphorylation. Use phosphatase inhibitors in control reactions [82].
- Auto-activation Phase: Incubate kinase (100 nM) with ATP (100 µM) and activating compounds (e.g., 1 mM CaCl₂ for CDPKs) for 30 minutes at 30°C [80].
- Substrate Phosphorylation Phase: Add substrate (e.g., 100 µM Syntide-2) and additional [γ-³²P]ATP (10 µM, 0.5 µCi/µL). Continue incubation for 15-60 minutes.
- Reaction Termination: Add SDS-PAGE loading buffer and heat at 95°C for 5 minutes.
- Detection: Separate proteins by SDS-PAGE, transfer to PVDF membrane, and analyze by autoradiography or phosphorimaging.
- Alternative Phospho-relay Detection: For kinases like IKK2, after auto-activation, add substrate with ADP instead of ATP to detect phosphate transfer from autophosphorylated kinase to substrate [23].
Troubleshooting:
- Include phosphorylation site mutants (e.g., T220N in CK1δ) as negative controls [82].
- Optimize auto-activation time and ATP concentration empirically.
- For tyrosine autophosphorylation detection, use anti-phosphotyrosine antibodies [23].
KiC Assay for Orphan Kinase Substrate Identification
Purpose: To identify putative substrates for orphan kinases using synthetic peptide libraries.
Materials:
- Purified orphan kinase (catalytic domain often sufficient)
- Synthetic peptide library (typically 15-20 amino acids with central phosphorylation sites)
- Kinase assay buffer
- ATP solution
- LC-MS/MS system with phosphopeptide enrichment capability
- Strong cation exchange (SCX) chromatography materials
- TiO₂ or IMAC phosphopeptide enrichment columns
Procedure:
- Peptide Library Design:
- Kinase Reaction: Incubate purified kinase (50-100 nM) with peptide library (10-50 µM each peptide) and ATP (100 µM) for 60 minutes at 30°C.
- Reaction Termination: Add trifluoroacetic acid (TFA) to 1% final concentration.
- Phosphopeptide Enrichment:
- Desalt peptides using C18 solid-phase extraction
- Enrich phosphopeptides using TiO₂ or IMAC chromatography [81]
- LC-MS/MS Analysis:
- Separate peptides by liquid chromatography
- Analyze by tandem mass spectrometry with data-dependent acquisition
- Data Analysis:
- Identify phosphorylated peptides using database search algorithms
- Quantify phosphorylation using spectral counting [81]
- Apply selection criteria (e.g., >2-fold enrichment over controls, presence in multiple replicates)
Validation: Confirm identified substrates through in vitro kinase assays with individual peptides and cellular validation using genetic approaches.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Challenging Kinase Studies
| Reagent/Category | Specific Examples | Function/Application | References |
|---|---|---|---|
| Generic Kinase Substrates | Casein (dephosphorylated), Myelin Basic Protein (MBP), Histones, Kemptide (LRRASLG), Syntide-2 (PLARTLSVAGLPGKK) | Initial activity screening for kinases with unknown specific substrates | [80] |
| Autophosphorylation Detection Tools | Lambda phosphatase, Anti-phosphotyrosine antibodies, Phospho-specific antibodies (e.g., anti-pT220 for CK1δ) | Characterizing autophosphorylation sites and stoichiometry | [23] [82] |
| Kinase Inhibitor Libraries | PKIS1/PKIS2 (Protein Kinase Inhibitor Sets), EMD Millipore inhibitor library | KiPIK screening, selectivity profiling, chemical validation | [83] |
| Computational Prediction Tools | MUsite, HMMER, PhoSiteformer, CSPred, NetworKIN | Predicting phosphorylation sites, kinase-substrate relationships, and critical regulatory sites | [81] [84] [10] |
| Peptide Library Resources | Custom synthetic peptide libraries, KiC assay platforms | High-throughput substrate identification for orphan kinases | [81] |
| Mass Spectrometry Platforms | LC-MS/MS with phosphopeptide enrichment (TiO₂, IMAC), Quantitative phosphoproteomics | Comprehensive identification and quantification of phosphorylation events | [81] [10] [82] |
Characterizing kinases with complex autophosphorylation requirements and orphan kinases remains challenging but essential for advancing kinase biology and drug discovery. The integrated approaches outlined here—combining traditional biochemistry with innovative screening methods and computational predictions—provide a roadmap for tackling these difficult targets. As kinase research evolves, several emerging technologies show particular promise: deep learning models for predicting critical phosphorylation sites [84], expanded kinase inhibitor libraries with comprehensive profiling data [83], and improved mass spectrometry-based methods for kinase activity inference [10]. These advances will accelerate our understanding of challenging kinase systems and facilitate the development of targeted therapies for diseases mediated by these important enzymes.
Ensuring Reagent Quality and Purity for Reproducible Results
In kinase activity and phosphorylation detection research, the quality of reagents is a foundational pillar supporting the integrity and reproducibility of experimental data. Reagents influence every stage of the workflow, from cell culture and protein extraction to phosphoprotein enrichment and analytical detection. Selecting the appropriate reagent purity grade is not merely a procedural formality but a critical scientific consideration that directly impacts assay sensitivity, specificity, and ultimately, the reliability of research conclusions. This application note provides a structured framework for selecting reagents based on their purity specifications and offers detailed protocols to ensure the generation of reproducible, high-quality data in kinase research.
Understanding Reagent Purity Grades
Reagent purity grades are standardized classifications that indicate a chemical's level of purity and its suitability for specific applications. These grades signify that a substance meets predetermined lower limits for specific chemical and biological impurities [86]. For biochemical assays focused on kinase activity, selecting the correct grade is paramount, as impurities can introduce experimental artifacts, inhibit enzymatic reactions, or cause false positives/negatives.
Classification of Common Purity Grades
The following table summarizes the most prevalent reagent grades encountered in biomedical research, their specifications, and their typical applications in kinase and phosphorylation studies:
Table 1: Common Reagent Purity Grades and Their Applications in Kinase Research
| Purity Grade | Defining Standards | Typical Purity | Suitable Applications in Kinase/Phosphorylation Research |
|---|---|---|---|
| ACS | American Chemical Society [87] | ≥95% [87] | Preparation of standard solutions for analytical assays; buffer components for kinetic studies. |
| Reagent | General analytical standards [87] | ≥95% [87] | General laboratory solutions, cell culture media supplements, and protein assay reagents. |
| USP/NF | United States Pharmacopeia–National Formulary [87] [86] | Meets pharmacopeial standards [87] | Suitable for processes aimed at therapeutic development, in vitro and in vivo assays. |
| Molecular Biology Grade | Absence of specific enzymes (e.g., DNases, RNases, proteases) [86] | Not specified by percentage | PCR, cloning, plasmid production for kinase expression constructs; critical for molecular biology steps. |
| HPLC Grade | Low UV absorbance and specific chemical purity [88] | High, with strict UV specs [88] | Liquid chromatography for phosphopeptide separation and analysis (e.g., LC-MS/MS). |
| Biotech/Biochemistry Grade | Tested for absence of inhibitors (e.g., heavy metals) [88] | High | Cell-based kinase assays, enzyme kinetics, preparation of biochemical assay buffers. |
For kinase research, ACS, Reagent, and USP-NF grades are often interchangeable and suitable for preparing most standard solutions and buffers [87] [86]. However, more specialized techniques require specific grades. For instance, Molecular Biology Grade is essential for PCR and other enzymatic reactions used in constructing kinase expression vectors, as contaminants can degrade enzymes and nucleic acids [86]. Similarly, HPLC Grade solvents are critical for chromatographic separations in mass spectrometry-based phosphoproteomics to avoid extraneous peaks that interfere with data interpretation [86].
A Framework for Reagent Selection
Choosing the correct reagent involves more than selecting a purity grade from a catalog. It requires a systematic evaluation of the experimental application, regulatory requirements, and supplier reliability.
Selection Criteria
- Application Compatibility: The primary consideration is whether the reagent grade is fit-for-purpose. For sensitive molecular biology techniques like PCR, which is routinely used in viral vector production for kinase studies, reagents must be free of DNase, RNase, and protease activity [86]. For cell culture, reagents must be sterile and endotoxin-free.
- Regulatory Compliance: Research with clinical or diagnostic applications must adhere to regulatory standards. The U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA) require a transition from Research Use Only (RUO) materials to those produced according to Good Manufacturing Practices (GMP) for commercial therapeutic manufacture [86]. Using high-quality, appropriate-grade reagents from the initial research stages can ease this transition.
- Supplier Reliability: Selecting a reputable supplier is integral to a successful workflow. Suppliers certified through International Organization for Standardization (ISO) inspections, such as ISO 13485:2016 for medical devices, typically have robust quality systems, including raw material analysis and bioburden testing [86].
Experimental Protocol: Qualification of New Reagent Batches
To ensure consistency and reproducibility, new reagent batches should be qualified before use in critical experiments.
Principle: Compare the performance of a new reagent batch against the current, validated batch in a standardized kinase activity assay.
Materials:
- Test Reagent: New batch of the reagent to be qualified.
- Control Reagent: Current, accepted batch of the same reagent.
- Active Kinase Enzyme (e.g., PKA, JAK2, MAPK).
- Specific Fluorescent or Luminescent Kinase Substrate.
- Reaction Buffer (e.g., 50 mM HEPES, pH 7.5, 10 mM MgCl₂, 1 mM DTT).
- ATP Solution.
- Stop Solution (compatible with detection method).
- Plate Reader (e.g., for fluorescence or luminescence).
Procedure:
- Prepare Reaction Mixtures: Using the test and control reagents, prepare two separate sets of kinase reaction buffers.
- Set Up Assay: In a 96-well plate, set up a serial dilution of the kinase enzyme for both the test and control buffer conditions.
- Initiate Reaction: To each well, add the substrate and ATP to start the enzymatic reaction. The final reaction volume is typically 50-100 µL.
- Incubate: Incubate the plate at 30°C for a predetermined time (e.g., 30 minutes).
- Stop Reaction: Add an equal volume of stop solution to each well.
- Detect Signal: Measure the output (e.g., fluorescence, luminescence) according to the substrate's specifications.
- Data Analysis: Plot the signal against the kinase concentration for both test and control conditions. Calculate the Z' factor, a statistical parameter for assay quality, for both batches. A Z' factor > 0.5 indicates an excellent assay. The performance of the test reagent batch is acceptable if the Z' factor is > 0.5 and the dose-response curves are not statistically different from the control (e.g., p > 0.05 in a Student's t-test comparing EC₅₀ values).
Reagent Management in a Kinase Research Workflow
The following diagram illustrates the critical decision points for reagent quality within a typical kinase and phosphorylation research workflow.
Diagram 1: Reagent quality checkpoints in a kinase research workflow.
The Scientist's Toolkit: Essential Reagent Solutions
The following table details key reagents and materials critical for success in kinase and phosphorylation research, along with their intended functions and recommended purity grades.
Table 2: Essential Research Reagent Solutions for Kinase and Phosphorylation Studies
| Reagent/Material | Function | Recommended Purity Grade |
|---|---|---|
| Water | Solvent for all aqueous solutions; mobile phase for LC-MS. | HPLC Grade for LC-MS; Molecular Biology Grade for enzymatic reactions; Ultra Pure for trace analysis [86] [88]. |
| Buffer Salts (e.g., Tris, HEPES) | Maintain physiological pH in assay buffers and cell culture media. | ACS Grade or higher to ensure accurate pH and low UV absorbance [87]. |
| Protease & Phosphatase Inhibitors | Prevent proteolytic degradation and dephosphorylation of target proteins during extraction. | Biotech/Biochemistry Grade to ensure function and absence of cross-inhibitors [88]. |
| Kinase Substrates (Peptide/Protein) | Act as phosphate acceptors in activity assays to measure kinase function. | >95% purity, confirmed by HPLC and mass spectrometry. |
| ATP | Phosphate donor for kinase enzymatic reactions. | Molecular Biology Grade or Ultra Pure to ensure optimal activity and low contaminant levels. |
| Chromatography Solvents (Acetonitrile, Methanol) | Mobile phase for phosphopeptide separation via liquid chromatography. | HPLC Grade or MS Grade for minimal UV absorbance and chemical interference [86] [88]. |
| Phospho-specific Antibodies | Enrich or detect specific phosphorylated proteins or sites in Western blot, ELISA. | Validated for specific application (e.g., Western blot, IHC); check species reactivity and phospho-site specificity. |
The path to reproducible and reliable results in kinase activity and phosphorylation research is paved with meticulous attention to reagent quality. By understanding purity grades, implementing a systematic selection and qualification process, and using high-quality materials at each step of the workflow, researchers can significantly reduce experimental variability and enhance the credibility of their scientific findings. As kinase research continues to drive drug discovery and fundamental biological understanding, a rigorous approach to reagent management remains an indispensable component of the scientific method.
Data Confidence: Validation, Comparative Analysis, and Translation to Complex Systems
In the field of kinase drug discovery, the reliability of biochemical assays directly impacts the success of identifying potent and selective inhibitors. Assay validation provides the critical foundation for high-quality research data and confident decision-making throughout the drug development pipeline. For researchers focusing on kinase activity and phosphorylation detection, three statistical parameters form the cornerstone of assay quality assessment: the Z'-factor, the Signal-to-Background ratio, and the Coefficient of Variation [89] [90]. These metrics collectively evaluate the robustness, dynamic range, and precision of experimental systems, enabling scientists to distinguish true biological signals from experimental noise [90]. This application note details the theoretical basis, calculation methods, and practical application of these parameters specifically for biochemical kinase assays, providing validated protocols to ensure data quality and reproducibility in high-throughput screening environments.
Theoretical Foundations of Key Assay Parameters
Z'-factor (Z-prime factor)
The Z'-factor is a statistical metric used to assess the quality and suitability of an assay, particularly in high-throughput screening (HTS) environments. It integrates both the dynamic range (separation between positive and negative controls) and the data variability (signal spread) into a single value [89] [90]. Unlike simpler metrics, the Z'-factor accounts for variability in both positive and negative control populations, providing a more realistic prediction of how an assay will perform when scaled to thousands of samples [90].
Calculation and Interpretation: The Z'-factor is defined by the following equation:
[ Z' = 1 - \frac{3(\sigmap + \sigman)}{|\mup - \mun|} ]
Where:
- μₚ = mean of positive control
- μₙ = mean of negative control
- σₚ = standard deviation of positive control
- σₙ = standard deviation of negative control [89] [90] [91]
The Z'-factor ranges from -∞ to 1, where a value of 1 represents an ideal assay with no variability, and values ≤ 0 indicate significant overlap between positive and negative controls, rendering the assay essentially useless for screening purposes [91]. The following table provides standard interpretation guidelines for Z'-factor values in HTS:
Table 1: Interpretation of Z'-factor Values
| Z'-factor Range | Assay Quality | Interpretation |
|---|---|---|
| 0.8 – 1.0 | Excellent | Ideal separation and low variability [90] |
| 0.5 – 0.8 | Good | Suitable for HTS [90] [91] |
| 0 – 0.5 | Marginal | Requires optimization [90] [91] |
| < 0 | Poor | Controls overlap; assay is unreliable [90] [91] |
It is important to note that while a Z'-factor > 0.5 is traditionally considered the threshold for HTS, recent research suggests this requirement may be overly stringent for certain assay formats, particularly cell-based or phenotypic assays with inherently higher biological variability [89] [92]. In such cases, assays with Z' < 0.5 may still provide valuable screening data when justified by biological necessity and supported by appropriate statistical thresholds [92].
Signal-to-Background Ratio (S/B)
The Signal-to-Background Ratio is a straightforward measure of assay window size, calculated as the ratio of the positive control signal to the negative control signal [90]. While simple to calculate and intuitive to understand, S/B has a significant limitation: it does not account for data variability [90].
Calculation: [ S/B = \frac{\mup}{\mun} ]
Where:
- μₚ = mean of positive control
- μₙ = mean of negative control [90]
Two assays can have identical S/B ratios yet perform very differently in screening due to differences in their signal variability. An assay with a high S/B but large variance may perform worse than an assay with a lower S/B but tight data distribution [90]. Therefore, S/B should never be used as a standalone metric for assay quality.
Coefficient of Variation (CV)
The Coefficient of Variation is a standardized measure of dispersion, expressing the standard deviation as a percentage of the mean [93]. It is particularly useful for comparing the degree of variation between datasets with different units or widely different means [93].
Calculation: [ CV = \frac{\sigma}{\mu} \times 100\% ]
Where:
In kinase assay validation, two types of CV are particularly relevant:
- Intra-assay CV: Measures precision within a single experiment (e.g., variation between replicate wells on the same plate) [94]. Acceptable intra-assay CVs are typically <10% [94].
- Inter-assay CV: Measures precision between different experimental runs (e.g., plate-to-plate or day-to-day variation) [94]. Acceptable inter-assay CVs are generally <15% [94].
Table 2: Summary of Key Assay Validation Parameters
| Parameter | Measures | Calculation | Acceptance Criterion | ||
|---|---|---|---|---|---|
| Z'-factor | Overall assay quality | ( 1 - \frac{3(\sigmap + \sigman)}{ | \mup - \mun | } ) | > 0.5 (HTS suitable) [90] [91] |
| S/B Ratio | Assay window size | ( \frac{\mup}{\mun} ) | Context-dependent; higher is better | ||
| Intra-assay CV | Within-plate precision | ( \frac{\sigma}{\mu} \times 100\% ) | < 10% [94] | ||
| Inter-assay CV | Plate-to-plate precision | ( \frac{\sigma}{\mu} \times 100\% ) | < 15% [94] |
Experimental Protocol: Validation of a Kinase Activity Assay
This protocol outlines the procedure for validating a biochemical kinase assay using ADP-Glo technology, a luminescence-based method that detects ADP formation as a measure of kinase activity.
Materials and Equipment
- Purified kinase enzyme (e.g., AMPKα1β1γ1 complex)
- Kinase substrate (specific peptide or protein)
- ATP solution
- ADP-Glo Kinase Assay Kit
- Assay buffer (e.g., 50 mM HEPES pH 7.5, 10 mM MgCl₂, 1 mM DTT)
- Reference inhibitor (e.g., staurosporine for ATP-competitive inhibitors)
- Specific inhibitor (e.g., BAY-3827 for AMPK)
- White, solid-bottom 384-well microplates
- Multichannel pipettes and reagent reservoirs
- Plate centrifuge
- Microplate reader capable of luminescence detection
- Data analysis software (e.g., GraphPad Prism, Microsoft Excel)
Reagent Preparation
- Kinase Solution: Prepare kinase enzyme in assay buffer at a concentration that yields linear reaction kinetics. For AMPK, a working concentration of 50 ng/mL (1 ng/well) has been validated [95].
- Substrate Solution: Prepare substrate in assay buffer. The concentration should be optimized based on the Km value for the specific kinase-substrate pair.
- ATP Solution: Prepare ATP in assay buffer. For inhibitor screening, use a concentration near the Km ATP (e.g., 3 × Km). For AMPK, 87 μM ATP has been used successfully [95].
- Control Solutions:
- Positive Control: Enzyme + substrate + ATP (maximum kinase activity)
- Negative Control 1: Substrate + ATP only (no enzyme)
- Negative Control 2: Enzyme + substrate + specific inhibitor (fully inhibited)
Assay Procedure
Plate Setup:
- Dispense 5 μL of assay buffer to all wells for background correction.
- Add 0.5 μL of reference inhibitor in DMSO to appropriate wells (final DMSO concentration ≤1%).
- Add 5.5 μL of kinase solution to all wells except negative control 1.
- Incubate for 15 minutes at room temperature.
Kinase Reaction:
- Add 2 μL of substrate solution to all wells.
- Initiate the reaction by adding 2 μL of ATP solution.
- Incubate for 30-60 minutes at room temperature (optimize for linear kinetics).
ADP Detection:
- Add 10 μL of ADP-Glo Reagent to all wells.
- Incubate for 40 minutes at room temperature to terminate kinase reaction and deplete remaining ATP.
- Add 20 μL of Kinase Detection Reagent to all wells.
- Incubate for 30-60 minutes at room temperature to convert ADP to ATP.
- Measure luminescence signal using a microplate reader.
Data Collection for Validation Parameters
For Z'-factor Calculation:
- Collect at least 16 replicate measurements for both positive and negative controls [90].
- Calculate means and standard deviations for both control populations.
- Compute Z'-factor using the formula in Section 2.1.
For S/B Ratio Calculation:
- Using the same control data, calculate the mean signal for positive controls divided by the mean signal for negative controls.
For CV Calculation:
- Intra-assay CV: Calculate the CV for replicate measurements within the same plate.
- Inter-assay CV: Repeat the assay on three separate days with freshly prepared solutions. Calculate the mean of control samples for each day, then determine the CV of these means.
Results and Data Analysis
Example Data Set and Calculations
The following table presents sample data from a validated AMPK kinase assay:
Table 3: Sample Validation Data from an AMPK Kinase Assay
| Parameter | Positive Control | Negative Control (No Enzyme) | Calculated Value |
|---|---|---|---|
| Mean Signal (RLU) | 120,000 | 12,000 | - |
| Standard Deviation | 8,000 | 1,200 | - |
| Z'-factor | - | - | 0.78 (Excellent) [95] |
| S/B Ratio | - | - | 10.0 |
| Intra-assay CV | 6.7% | 10.0% | - |
| Inhibitor IC₅₀ (Staurosporine) | - | - | 0.45 nM [95] |
Sample Calculation: [ Z' = 1 - \frac{3(8,000 + 1,200)}{|120,000 - 12,000|} = 1 - \frac{3(9,200)}{108,000} = 1 - \frac{27,600}{108,000} = 0.78 ]
Troubleshooting and Optimization
Low Z'-factor ( < 0.5):
- If σₚ ≫ σₙ: High signal variability - optimize enzyme concentration, incubation time, or reagent stability.
- If σₙ ≫ σₚ: High background variability - improve washing steps, reduce background signal, or use purer reagents.
- If |µₚ–µₙ| is small: Small assay window - increase substrate concentration, optimize ATP levels, or use a more sensitive detection method [90].
High Intra-assay CV ( > 10%):
- Check pipetting technique and calibrate pipettes.
- Pre-wet pipette tips before dispensing viscous solutions.
- Ensure complete mixing of reagents without generating bubbles.
- Centrifuge plates before reading to remove bubbles [94].
High Inter-assay CV ( > 15%):
- Prepare fresh reagent aliquots for each experiment.
- Standardize incubation times and temperatures.
- Use the same reagent lots throughout the validation.
- Include quality control samples on each plate.
Research Reagent Solutions for Kinase Assays
Table 4: Essential Research Reagents for Kinase Activity Assays
| Reagent / Technology | Function | Example Applications |
|---|---|---|
| HTRF KinEASE | TR-FRET-based detection of phosphorylated peptides | Tyrosine kinase assays (e.g., Syk kinase) [96] |
| ADP Hunter / ADP-Glo | Luminescence-based ADP detection | Universal kinase activity screening [95] |
| Transcreener ADP Assays | Fluorescence polarization-based ADP detection | High-throughput kinase inhibitor screening [90] |
| HitHunter Kinase Activity Kits | Fluorescence-based kinase activity detection | Kinase inhibitor profiling [95] |
| AlphaLISA / AlphaPlex | Bead-based proximity assays | Simultaneous detection of multiple kinase targets [89] |
Visualizations
Assay Validation Workflow
Statistical Relationship Between Key Parameters
Robust assay validation is a prerequisite for successful kinase research and inhibitor screening. The complementary parameters of Z'-factor, Signal-to-Background ratio, and Coefficient of Variation provide a comprehensive framework for assessing assay quality and reliability. By implementing the protocols and guidelines outlined in this application note, researchers can ensure their kinase activity assays generate pharmacologically relevant data, ultimately accelerating the discovery of novel therapeutic agents. While traditional thresholds for these parameters provide useful benchmarks, they should be applied with scientific judgment, particularly for complex biological systems where some variability is inherent.
Kinase activity and phosphorylation detection represent a cornerstone of modern biochemical research, particularly in drug discovery and signal transduction studies. Protein kinases regulate virtually all cellular processes, and their dysregulation is implicated in a vast array of diseases, including cancer, autoimmune disorders, and neurodegenerative conditions [97] [2]. The global kinase drug discovery solutions market reflects this importance, demonstrating steady growth driven by increasing cancer incidence, focus on precision medicine, and adoption of advanced screening technologies [98]. Within this market, fluorescence-based detection and analysis is witnessing higher growth due to superior sensitivity and faster analysis times compared to traditional methods [98].
The fundamental biochemical reaction underlying all kinase assays involves the transfer of a phosphate group from adenosine triphosphate (ATP) to serine, threonine, or tyrosine residues on substrate proteins, generating adenosine diphosphate (ADP) as a byproduct [2]. Detection technologies have evolved to measure either substrate phosphorylation or ADP formation, each with distinct advantages and limitations. This application note provides a comprehensive comparative analysis of major detection platforms, emphasizing practical implementation for researchers navigating the complex landscape of kinase assay technologies.
Technology Comparison and Performance Metrics
The selection of an appropriate detection technology requires careful consideration of performance metrics aligned with experimental goals. Key parameters include sensitivity, throughput, cost-effectiveness, and universality across different kinase targets.
Table 1: Comprehensive Comparison of Major Kinase Detection Technologies
| Technology | Detection Principle | Sensitivity | Throughput | Cost per Sample | Key Advantages | Primary Limitations |
|---|---|---|---|---|---|---|
| Radiometric | Measures incorporation of ³²P from [γ-³²P]ATP into substrates | Very High (historical "gold standard") | Low to Medium | Low (reagents) / High (waste disposal) | Direct measurement; minimal substrate modification | Radioactive hazards; specialized disposal requirements |
| Antibody-Based | Detects phosphorylation using phospho-specific antibodies | High (depends on antibody quality) | Medium to High | Medium to High | Site-specific information; well-established protocols | Antibody-dependent; limited universality; potential cross-reactivity |
| Luminescent (e.g., ADP-Glo) | Converts ADP to ATP with luciferase-generated luminescence | High | High | Medium | Homogeneous format; good sensitivity | Multi-step protocol; coupling enzyme-dependent |
| Fluorescence-Based ADP Detection (e.g., Transcreener) | Directly measures ADP via fluorescence polarization (FP) or TR-FRET | High | Very High (384-/1536-well) | Low to Medium | Universal format; mix-and-read simplicity; minimal interference | Requires fluorescence-capable plate reader |
| FRET-Based Biosensors (e.g., Picchu-B) | Monitors kinase activity via conformation-dependent FRET changes | Medium to High | Medium | Low (after initial development) | Real-time kinetics; applicable in live cells | Requires sensor engineering and optimization |
| Mass Spectrometry-Based (e.g., ProKAS) | Quantifies phosphorylated peptides via mass spectrometry | Very High | Low to Medium (increasing with automation) | High | Multiplexing capability; unambiguous site identification | Specialized instrumentation; complex data analysis |
Recent technological advances have addressed longstanding limitations in kinase activity monitoring. Fluorescence-based ADP detection assays, such as the Transcreener platform, have gained prominence due to their universal applicability—they work with any kinase and substrate combination by measuring the common ADP product [97]. This universality eliminates the need for kinase-specific reagents, making these assays particularly valuable for high-throughput screening (HTS) campaigns and structure-activity relationship studies during lead optimization [97]. The homogeneous, mix-and-read format significantly reduces hands-on time while providing robust performance with Z' values ideal for HTS applications [97].
Emerging technologies continue to push the boundaries of what's possible in kinase research. The Proteomic Kinase Activity Sensor (ProKAS) technique, recently described in Nature Communications, represents a significant innovation by enabling multiplexed, spatially resolved monitoring of kinase activity using mass spectrometry [5]. This system employs a tandem array of peptide sensors with amino acid barcodes that allow simultaneous tracking of multiple kinases within a single polypeptide module, overcoming the multiplexing limitations of fluorescence-based biosensors caused by spectral overlap [5]. Meanwhile, FRET-based biosensors like Picchu-B have been successfully implemented for real-time monitoring of specific kinases such as EGFR and its clinically relevant mutants, offering insights into kinetic parameters and inhibitor profiling in both cellular and in vitro contexts [99].
Application Notes & Experimental Protocols
Protocol: Universal ADP Detection Assay for High-Throughput Screening
Principle: This homogeneous, mix-and-read assay directly quantifies ADP production using fluorescence polarization (FP) or TR-FRET detection, applicable to any kinase-substrate combination [97].
Reagents and Solutions:
- Kinase reaction buffer (optimized for specific kinase)
- ATP solution (variable concentration for KM determination)
- Test compounds in DMSO (final concentration typically <1%)
- Purified kinase enzyme
- Substrate (peptide or protein)
- Transcreener ADP₂ Detection Mix (containing antibody and tracer)
- Stop solution
Procedure:
- Reaction Setup: In a low-volume 384-well or 1536-well plate, add 2 µL of compound solution or DMSO control to appropriate wells.
- Enzyme/Substrate Addition: Add 4 µL of kinase-substrate mixture prepared in reaction buffer. For negative controls, include wells without enzyme or without substrate.
- Reaction Initiation: Initiate kinase reaction by adding 4 µL of ATP solution prepared in reaction buffer. Final reaction volume is 10 µL.
- Incubation: Incubate plate at room temperature or 30°C for appropriate time (typically 30-120 minutes) to ensure linear initial velocity conditions.
- Detection: Add 10 µL of Transcreener Detection Mix containing ADP-selective antibody and fluorescent tracer. Incubate for 30-60 minutes.
- Readout: Measure FP or TR-FRET signal using compatible plate reader.
- Data Analysis: Calculate percentage inhibition using positive (no compound) and negative (no enzyme) controls. Determine IC₅₀ values using nonlinear regression analysis.
Critical Considerations:
- Z' Factor Validation: Ensure robust assay performance by calculating Z' factor >0.5 using positive and negative controls.
- ATP Concentration: Use ATP concentrations near the KM value for the kinase of interest to maximize sensitivity to inhibitors.
- DMSO Tolerance: Verify that final DMSO concentration (typically ≤1%) does not affect kinase activity or detection.
- Signal Stability: Confirm signal stability over measurement period, as FP/TR-FRET signals are generally stable for several hours.
Protocol: FRET-Based Kinase Activity Monitoring with Picchu-B Biosensor
Principle: This protocol utilizes the Picchu-B recombinant biosensor, a single polypeptide incorporating FRET pairs that undergoes conformational changes upon phosphorylation, enabling real-time kinase activity monitoring [99].
Reagents and Solutions:
- Recombinant Picchu-B biosensor (expressed and purified from bacterial system)
- Kinase reaction buffer (50 mM HEPES, pH 7.5, 10 mM MgCl₂, 1 mM EGTA)
- ATP solution (prepared fresh in reaction buffer)
- Purified EGFR kinase domain (wild-type or mutant)
- Reference inhibitor (e.g., Erlotinib for EGFR)
Procedure:
- Biosensor Validation: Confirm biosensor concentration and functionality by measuring basal FRET ratio in the absence of kinase.
- Reaction Setup: In a black-walled 96-well or 384-well plate, add 40 µL of Picchu-B solution (optimal concentration determined by titration, typically 0.5-2 µM).
- Baseline Measurement: Acquire initial FRET measurements (excitation: 433 nm, emission: 475 nm and 527 nm) to establish baseline.
- Reaction Initiation: Add 10 µL of EGFR kinase solution followed by 10 µL of ATP solution to initiate reaction. For inhibitor studies, pre-incubate kinase with compounds for 15 minutes before ATP addition.
- Real-Time Monitoring: Continuously monitor FRET ratio (527 nm/475 nm emission) using a plate reader equipped with temperature control and kinetic capability.
- Data Collection: Acquire readings every 30-60 seconds for 60-120 minutes.
- Kinetic Analysis: Calculate initial velocities from the linear portion of the progress curve. Determine IC₅₀ values by fitting inhibitor dose-response data to appropriate model.
Critical Considerations:
- Biosensor Specificity: Validate biosensor specificity using kinase-deficient mutants or selective inhibitors.
- Signal-to-Noise Optimization: Optimize biosensor concentration to maximize dynamic range while minimizing background.
- Linear Range Determination: Establish the linear range of the assay with respect to time and enzyme concentration.
- Interference Assessment: Test compounds for intrinsic fluorescence that might interfere with FRET measurements.
Protocol: Multiplexed Kinase Activity Profiling with ProKAS
Principle: The Proteomic Kinase Activity Sensor technique employs barcoded peptide sensors in a tandem array for multiplexed kinase activity monitoring via mass spectrometry, enabling spatial resolution and high-content screening [5].
Reagents and Solutions:
- ProKAS construct with targeting elements (NLS, NES, or specific organelle tags)
- transfection reagent suitable for target cell line
- Cell lysis buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% NP-40, protease and phosphatase inhibitors)
- ALFA tag affinity resin for purification
- Trypsin digestion solution
- LC-MS/MS solvents (0.1% formic acid in water and acetonitrile)
Procedure:
- Sensor Expression: Transfect cells with ProKAS construct using optimized protocol for specific cell line.
- Treatment: Apply experimental treatments (e.g., kinase inhibitors, growth factors, genotoxic agents) for predetermined times.
- Cell Lysis: Harvest cells and lyse in appropriate buffer. Clarify lysates by centrifugation at 16,000 × g for 15 minutes.
- Affinity Purification: Incubate cleared lysates with ALFA tag affinity resin for 1-2 hours at 4°C. Wash extensively with lysis buffer.
- On-Bead Digestion: Digest purified ProKAS sensor with sequencing-grade trypsin overnight at 37°C.
- LC-MS/MS Analysis: Analyze resulting peptides using targeted LC-MS/MS method (e.g., parallel reaction monitoring) focused on barcoded sensor peptides.
- Data Processing: Quantify both phosphorylated and unphosphorylated forms of each sensor peptide. Calculate phosphorylation ratio as (phosphopeptide)/(phosphopeptide + unmodified peptide).
Critical Considerations:
- Sensor Design: Incorporate flanking arginine residues to ensure proper tryptic cleavage and MS detection.
- Barcode Selection: Use unique amino acid barcodes with minimal natural occurrence to avoid background interference.
- Targeting Validation: Confirm proper subcellular localization of ProKAS constructs using fluorescence microscopy.
- MS Optimization: Develop optimized LC-MS/MS methods for each sensor peptide to maximize sensitivity and reproducibility.
The Scientist's Toolkit: Essential Research Reagent Solutions
Successful implementation of kinase activity assays requires careful selection of appropriate reagents and tools. The following table summarizes key solutions and their applications:
Table 2: Essential Research Reagents for Kinase Activity and Phosphorylation Detection
| Reagent Category | Specific Examples | Function & Application | Key Considerations |
|---|---|---|---|
| Universal Detection Kits | Transcreener ADP² Assay, ADP-Glo | Detect ADP formation as universal kinase reaction product | Compatibility with substrate type; HTS compatibility; signal stability |
| Biosensor Systems | Picchu-B, FRET-based kinase biosensors | Real-time monitoring of specific kinase activities in vitro and in live cells | Specificity validation; dynamic range; expression efficiency |
| Multiplexed Platforms | ProKAS, Peptide Arrays | Simultaneous monitoring of multiple kinase activities | MS compatibility; barcode design; spatial targeting efficiency |
| Phospho-Specific Antibodies | Anti-phospho-S175-SnRK2, Phospho-tyrosine antibodies | Selective detection of specific phosphorylation events | Specificity validation; application compatibility (Western, ICC) |
| Kinase Inhibitor Libraries | Selective and pan-kinase inhibitors | Tool compounds for pathway validation and mechanism studies | Selectivity profile; potency; solvent compatibility |
| Specialized Substrates | Histone H3, myelin basic protein, synthetic peptides | Optimal substrates for specific kinase families | KM values; phosphorylation efficiency; detection compatibility |
Workflow and Technology Selection Diagrams
Technology Selection Decision Pathway
ProKAS Experimental Workflow
The evolving landscape of kinase detection technologies offers researchers an expanding toolkit for investigating phosphorylation-dependent signaling networks. The optimal technology selection depends critically on experimental priorities: fluorescence-based ADP detection assays provide the strongest combination of universality, simplicity, and HTS compatibility for drug discovery applications [97]; FRET-based biosensors enable real-time kinetic analysis of specific kinase targets [99]; while emerging mass spectrometry-based approaches like ProKAS offer unprecedented multiplexing capabilities and spatial resolution [5].
Future directions in kinase activity monitoring will likely focus on enhancing spatial resolution in live cells, increasing multiplexing capacity, and integrating kinase activity data with other omics datasets. The ongoing development of more sensitive, reproducible, and accessible platforms will continue to advance our understanding of kinase biology and accelerate the discovery of novel therapeutic agents targeting dysregulated kinase signaling in human disease.
In targeted drug discovery, the journey from initial compound screening to a effective therapeutic hinges on a critical step: reliably predicting how a molecule's activity in a test tube (biochemical efficacy) translates to its desired effect in a living cell (cellular efficacy). This correlation is particularly crucial for kinase targets, as kinases regulate essential cellular processes like signaling, growth, and metabolism, and their dysregulation is implicated in cancer, metabolic, and neurodegenerative disorders [1]. Establishing a robust bridge between these two domains ensures that promising in vitro data accurately forecasts biological relevance, de-risking the drug development pipeline and accelerating the identification of viable clinical candidates.
The central challenge lies in the fundamental differences between purified enzyme systems and the complex cellular environment. A potent inhibitor in a biochemical assay may fail in a cellular context due to factors like cell permeability, efflux by transporters, compound metabolism, or competing ATP concentrations. This document provides detailed application notes and protocols designed to systematically address these challenges, enabling researchers to generate reliable and predictive data for kinase inhibitor development.
Application Notes: Core Concepts and Workflows
The Critical Role of Kinase Assays in Drug Discovery
Kinases are enzymes that catalyze the transfer of phosphate groups from adenosine triphosphate (ATP) to specific protein substrates, a process known as phosphorylation. This action serves as a primary regulatory mechanism for numerous cellular functions [1]. Kinase-targeted therapies, exemplified by imatinib, have revolutionized cancer treatment, but their application now extends to cardiovascular, autoimmune, and neurological research [1].
Biochemical and cell-based assays serve distinct but complementary purposes:
- Biochemical Assays: Utilize purified kinase enzymes to measure the direct impact of a compound on the kinase's catalytic activity. They are ideal for high-throughput screening (HTS) and mechanistic studies because they provide a controlled system free from cellular complexities.
- Cell-Based Assays: Evaluate a compound's functional effect in a live-cell context, measuring downstream events like pathway modulation, proliferation, or apoptosis. They confirm that a compound not only binds its target but also achieves sufficient intracellular concentration and modulates the intended biology.
Market and Technological Trends
The global market for cell-based assay services, a key component of this pipeline, is experiencing significant growth, projected to reach approximately $2,500 million by 2025 with a robust Compound Annual Growth Rate (CAGR) of around 12.5% [100]. This expansion is fueled by escalating demand from biopharmaceutical and biotechnology companies for advanced drug discovery services and the increasing complexity of therapeutic targets [100]. Key trends include the adoption of high-throughput screening (HTS), multiplexing capabilities, and the growing outsourcing of these specialized assays to contract research organizations (CROs) [100].
Establishing Correlation through Stressed Samples
A powerful strategy for validating the bridge between in vitro and cell-based potency involves using "stressed samples." This method entails intentionally degrading a reference compound or biological agent (e.g., an mRNA vaccine or protein antigen) under controlled stress conditions, such as thermal stress, to create a series of samples with a gradient of potencies [101]. These samples are then tested in parallel in both biochemical/cell-based expression assays and in vivo immunogenicity or efficacy models.
A seminal example involves mRNA vaccine samples subjected to gradual thermal destabilization. The percentage of intact mRNA and subsequent in vitro protein expression were measured, demonstrating a direct correlation between structural integrity, in vitro potency, and in vivo immunogenicity [101]. This approach confirms that the in vitro assay is sensitive enough to detect changes that impact biological function, thereby validating its use as a predictive tool for lot release and stability testing, reducing reliance on variable and costly animal studies [101].
Table 1: Key Assay Formats for Kinase Activity Analysis
| Assay Category | Technology/Format | Key Principle | Best Use Cases |
|---|---|---|---|
| Biochemical Activity | Luminescence (e.g., ADP-Glo) | Quantifies ADP formation resulting from kinase reaction [1] | HTS, primary screening |
| Fluorescence (TR-FRET, FP) | Uses fluorescent labels to monitor binding or phosphorylation [1] | Binding affinity, competition studies | |
| Mobility Shift Assays | Separates phosphorylated from non-phosphorylated substrates [1] | Direct, quantitative activity measurement | |
| Cellular Efficacy | Cell Viability/Proliferation | Measures ATP content (e.g., via ATP Content Determination Method) as indicator of cell health and number [100] | Anti-cancer compound efficacy, toxicity |
| Phospho-Specific Flow Cytometry | Detects intracellular phosphorylation targets via fluorescent antibodies | Pathway modulation, pharmacodynamics | |
| High-Content Imaging | Multiparametric analysis of cell morphology and biomarker localization | Complex phenotypic screening | |
| Kinase Activity Inference | Phosphoproteomics with Computational Inference (e.g., KSEA, PTM-SEA) | Infers kinase activity from phosphoproteomics data using kinase-substrate libraries [10] | Systems biology, identifying dysregulated kinases in disease |
Experimental Protocols
Protocol 1: Biochemical Kinase Inhibition Assay (TR-FRET Format)
This protocol details a homogeneous, non-radioactive method for measuring kinase inhibition using Time-Resolved Förster Resonance Energy Transfer (TR-FRET), suitable for HTS.
3.1.1 Principle A fluorescently labeled substrate is phosphorylated by the kinase. A terbium-coupled anti-phospho-substrate antibody binds the phosphorylated product, bringing the donor (Tb) in close proximity to the acceptor (fluorescent dye on the substrate), generating a FRET signal. Inhibitors reduce phosphorylation, thereby decreasing the FRET signal [1].
3.1.2 Reagents and Materials
- Kinase Enzyme: Purified, recombinant human kinase of interest.
- ATP: Adenosine triphosphate, diluted to desired concentration in reaction buffer.
- Substrate: A peptide derived from a known physiological substrate, labeled with a suitable fluorophore (e.g., FITC).
- TR-FRET Detection Reagent: Contains anti-phospho-substrate antibody conjugated to terbium cryptate.
- Assay Buffer: Typically a mixture of HEPES (pH 7.4), MgCl₂, DTT, and BSA.
- Test Compounds: Dissolved in DMSO. Final DMSO concentration in the assay should be normalized (typically ≤1%).
- Low-Volume Assay Plates: White, 384-well plates.
3.1.3 Procedure
- Plate Preparation: Dispense 50 nL of compound in DMSO or DMSO control into assay plates using an acoustic dispenser or pintool.
- Enzyme/Substrate Mixture: Prepare a master mix containing kinase and substrate in assay buffer. Add 5 µL of this mix to each well.
- Reaction Initiation: Add 5 µL of ATP in assay buffer to each well to start the reaction. Final assay volume is 10 µL.
- Incubation: Cover the plate and incubate at room temperature for 60 minutes.
- Detection: Add 5 µL of TR-FRET detection reagent (prepared in an EDTA-containing buffer to stop the kinase reaction). Incubate for 30-60 minutes.
- Reading: Measure the TR-FRET signal on a compatible plate reader (e.g., excitation: 340 nm, emission: 495 nm & 520 nm). The ratio of 520 nm/495 nm emission is proportional to the amount of phosphorylated substrate.
3.1.4 Data Analysis
- Calculate % Inhibition for each compound:
[1 - (Ratio_compound - Ratio_min)/(Ratio_max - Ratio_min)] * 100Ratio_max: Average signal from DMSO control (no inhibition).Ratio_min: Average signal from control with no enzyme (full inhibition).
- Generate dose-response curves and calculate IC₅₀ values using non-linear regression (e.g., four-parameter logistic fit).
Protocol 2: Cell-Based Kinase Inhibition Assay (Phospho-Flow Cytometry)
This protocol measures the inhibition of a specific kinase target in cells by quantifying the phosphorylation levels of its direct downstream substrate using intracellular staining and flow cytometry.
3.2.1 Principle Cells are treated with compounds, fixed, and permeabilized. The phosphorylation status of the target protein is detected using a phospho-specific primary antibody, followed by a fluorescently labeled secondary antibody, or directly with a labeled primary antibody. The median fluorescence intensity (MFI) of the cell population, measured by flow cytometry, correlates with the level of phosphorylation and thus, kinase activity.
3.2.2 Reagents and Materials
- Cell Line: Chosen based on relevance to the kinase target and disease context. The HepG2 cell line, for instance, has been selected in optimized assays for protein expression from mRNA constructs due to its strong performance across multiple criteria [101].
- Stimulus: A cytokine, growth factor, or other agent known to activate the kinase pathway of interest (if required).
- Phospho-Specific Antibody: Antibody specific to the phosphorylated form of the kinase substrate.
- Fluorophore-Conjugated Secondary Antibody (if needed).
- Fixation/Permeabilization Buffer: Commercially available kits (e.g., BD Cytofix/Cytoperm) are recommended.
- Flow Cytometry Staining Buffer: PBS containing a low concentration of BSA or FBS.
- DMSO, for compound dilution.
3.2.3 Procedure
- Cell Seeding and Treatment: Seed cells in a 96-well plate. After adherence, pre-treat cells with serially diluted compounds for a predetermined time (e.g., 1-2 hours).
- Stimulation: If applicable, stimulate cells with the activating agent for a specific duration (e.g., 15-30 minutes) to induce phosphorylation.
- Fixation and Permeabilization: Rapidly transfer the plate to a centrifuge, aspirate media, and resuspend cells in fixation buffer. Incubate for 10-15 minutes at 37°C. Wash cells once with permeabilization buffer.
- Intracellular Staining: Resuspend cell pellets in permeabilization buffer containing the titrated phospho-specific antibody. Incubate for 30-60 minutes at room temperature in the dark.
- Washing and Analysis: Wash cells twice with staining buffer. Resuspend in a fixed volume of staining buffer and acquire data on a flow cytometer.
- Gating Strategy: Gate on viable cells based on forward and side scatter. Analyze the median fluorescence intensity (MFI) of the phospho-protein channel for the viable cell population.
3.2.4 Data Analysis
- Normalize MFI values to vehicle control (0% inhibition) and a control where pathway stimulation is omitted (100% inhibition).
- Plot % Inhibition vs. compound concentration to generate dose-response curves and calculate IC₅₀ values.
Table 2: Troubleshooting Common Issues in Kinase Assays
| Problem | Potential Cause | Suggested Remedy |
|---|---|---|
| High Background in Biochemical Assay | Non-specific binding, compound auto-fluorescence [1] | Include control wells without enzyme and without substrate; use low-fluorescence plates; test compounds for interference. |
| Poor Z'-factor in HTS | High variability, signal drift | Ensure reagent stability and equilibration to room temperature; use precision liquid handlers; check dispenser accuracy. |
| Lack of Correlation between Biochemical and Cellular IC₅₀ | Poor cell permeability, efflux pumps, compound metabolism/instability [1] | Assess cell permeability (e.g., P-gp assay); measure intracellular compound concentration; use a cell-permeable positive control. |
| Low Signal in Phospho-Flow Assay | Inefficient stimulation, antibody issues, over-fixation | Titrate stimulus concentration and time; titrate antibody; optimize fixation/permeabilization time and temperature. |
| High Variability in Cell-Based Data | Inconsistent cell seeding, treatment, or staining | Use automated plate washers; ensure homogeneous cell suspension during seeding and staining; include technical replicates. |
Advanced Correlation and Computational Inference
Leveraging Phosphoproteomics for Kinase Activity Inference
Beyond targeted assays, phosphoproteomics provides a systems-level view for correlating biochemical inhibition with global cellular changes. Mass spectrometry-based phosphoproteomics can identify and quantify thousands of phosphorylation sites, creating a snapshot of kinase and phosphatase activities within the cell [10].
Computational methods infer kinase activity from this data using kinase-substrate libraries. These methods, including Kinase-Substrate Enrichment Analysis (KSEA) and PTM-Signature Enrichment Analysis (PTM-SEA), analyze whether the known substrates of a particular kinase are significantly more phosphorylated or dephosphorylated than the background of all measured phosphosites [10]. This approach can identify not only the direct target but also downstream kinases affected by the inhibitor, providing a comprehensive view of pathway modulation.
Recent evaluations, such as those conducted by the benchmarKIN R package, highlight that while many computational methods perform similarly, the choice of kinase-substrate library (e.g., PhosphoSitePlus, SIGNOR) significantly impacts the quality of inferred activities. Combining manually curated libraries with predicted interactions from tools like NetworKIN can enhance coverage and performance [10].
Table 3: Key Research Reagent Solutions for Kinase Assay Development
| Reagent / Resource | Function / Description | Example(s) / Notes |
|---|---|---|
| Purified Recombinant Kinases | Essential reagent for biochemical assay development. Provides a controlled system for measuring direct compound binding and inhibition. | Available from various vendors (e.g., Thermo Fisher, Merck). Activity and purity should be validated. |
| Validated Cell Lines | Provide the cellular context for efficacy and permeability assessment. | Select lines with high endogenous target expression or engineered lines (e.g., overexpression, reporter systems). HepG2 is noted for high protein expression in mRNA transfection [101]. |
| Phospho-Specific Antibodies | Detect and quantify specific phosphorylation events in cell-based assays (e.g., Western blot, flow cytometry, immunofluorescence). | Critical for measuring pathway modulation. Must be rigorously validated for specificity. |
| Kinase-Substrate Libraries | Databases of known and predicted kinase-substrate relationships. Fuel computational inference of kinase activity from phosphoproteomic data. | PhosphoSitePlus, SIGNOR, Phospho.ELM. Performance varies; combined libraries often yield superior results [10]. |
| Advanced Detection Kits | Enable sensitive, homogeneous, and non-radioactive detection of kinase activity in HTS formats. | ADP-Glo, IMAP, LanthaScreen, HTRF kits. Offer robustness and scalability. |
| Benchmarking Datasets & Tools | Provide a gold standard for evaluating the performance of kinase activity inference methods. | The benchmarKIN R package uses perturbation experiments and tumor multi-omics data for comprehensive evaluation [10]. |
Correlating biochemical activity with cellular efficacy is not a single experiment but an iterative process that leverages multiple orthogonal techniques. The protocols and application notes detailed herein—from foundational biochemical and cell-based assays to advanced phosphoproteomic inference—provide a structured framework for building this critical bridge. The consistent use of stressed samples to create a potency gradient is a powerful strategy for formally validating this correlation. Furthermore, the integration of computational methods for kinase activity inference from global phosphoproteomic data offers a systems-level perspective, ensuring that in vitro potency data is consistently and reliably translated into predictable cellular and, ultimately, therapeutic outcomes. By systematically applying these principles, researchers can significantly enhance the efficiency and success rate of kinase-directed drug discovery programs.
Kinase inhibitor selectivity, defined as the tendency of a compound to bind to its intended kinase target over other off-target kinases, is a critical parameter in drug discovery and development. The high degree of structural conservation across the ATP-binding sites of the approximately 500 members of the human kinome makes achieving selectivity a formidable challenge [102]. A compound's selectivity profile directly influences both its therapeutic efficacy and its safety; unintended off-target binding can lead to severe adverse events and dose-limiting toxicities, though in some cases, it can also reveal opportunities for polypharmacology or drug repurposing [102] [103]. Consequently, robust and comprehensive selectivity profiling is indispensable for guiding lead optimization, assessing clinical safety, and understanding the mechanisms of drug action [103] [104].
This application note provides a detailed framework for assessing kinase inhibitor specificity, integrating both in silico and experimental approaches. We summarize key computational and statistical metrics for analyzing profiling data and present step-by-step protocols for major experimental platforms, providing researchers with a consolidated resource for kinome-wide selectivity assessment.
Computational and Statistical Approaches for Selectivity Analysis
Computational methods offer a rapid and cost-effective means for initial kinase inhibitor selectivity profiling across a wide kinome space, overcoming the limitations of available biochemical assays [102].
Binding Site Similarity Analysis with KinomeFEATURE
The KinomeFEATURE database enables kinase binding site similarity search by comparing protein microenvironments characterized using diverse physiochemical descriptors [102]. The method involves:
- Database Construction: The database comprises ~2850 kinase structures from 189 unique human kinases.
- Microenvironment Characterization: Protein microenvironments are composed of diverse physiochemical descriptors in 6-radial shells at binding residues of the co-crystal ligand.
- Similarity Scoring: The pocket similarity between two kinases is quantified using a PocketFEATURE score (PFS), which is based on the presence or absence of shared protein microenvironments. This approach does not necessitate a strong geometric requirement and is robust against protein conformational changes [102].
In a validation study, this method achieved over 90% accuracy in predicting the selectivity of 15 known kinase inhibitors and identified unexpected inhibitor cross-activities, such as between PKR and FGFR2 kinases [102].
Selectivity Metrics for Profiling Data Analysis
Once experimental profiling data is generated, several metrics can quantify selectivity. The following table summarizes key selectivity metrics used in kinase research.
Table 1: Key Metrics for Quantifying Kinase Inhibitor Selectivity
| Metric Name | Formula/Definition | Interpretation | Advantages and Limitations |
|---|---|---|---|
| Standard Selectivity Score (S(x)) | ( S(x) = \frac{\text{number of values} \geq x}{\text{total number of values}} ) [103] | A low S(x) indicates high selectivity; a high S(x) indicates poor selectivity. | Advantage: Simple, quantitative, and easily comparable.Limitation: Highly dependent on the chosen threshold (x) and does not capture nuances of affinity strength [103]. |
| Window Score (WS) | Not a simple formula; based on the difference in activity between primary and off-targets [103]. | Defines the selectivity window based on the potency drop-off between the primary target and other kinases. | Advantage: Easy to compute; offers a different viewpoint for compound selection [103]. |
| Ranking Score (RS) | Not a simple formula; based on the rank order of kinase targets by potency [103]. | Ranks kinases based on inhibitor potency, providing a profile of relative affinities. | Advantage: Easy to compute; offers a different viewpoint for compound selection [103]. |
| Gini Coefficient | A measure of statistical dispersion calculated from the Lorenz curve of inhibition data [103]. | Ranges from 0 (perfectly non-selective) to 1 (perfectly selective). | Advantage: A single number summarizing the entire selectivity profile.Limitation: Can be insensitive to a small number of strong off-target interactions [103]. |
| Selectivity Entropy | Based on the Shannon entropy of the inhibition profile [103]. | A higher entropy indicates a more promiscuous inhibitor. | Advantage: Another method to summarize the entire profile into one value [103]. |
Experimental Protocols for Selectivity Profiling
A multi-faceted approach using various assay technologies provides the most comprehensive view of inhibitor selectivity.
Biochemical Kinase Activity and Binding Assays
Biochemical assays measure the direct interaction between an inhibitor and a purified kinase enzyme, providing a clear assessment of intrinsic binding and inhibition.
Table 2: Comparison of Biochemical Kinase Profiling Assay Platforms
| Assay Platform | Detection Method | Measured Parameter | Key Features |
|---|---|---|---|
| Z'-LYTE / Adapta (Thermo Fisher) | Fluorescence / Time-Resolved FRET | Percentage Inhibition; IC~50~ | Measures peptide phosphorylation (Z'-LYTE) or ADP production (Adapta) for activity assessment [102]. |
| LanthaScreen (Thermo Fisher) | Fluorescence Polarization | Percentage Inhibition; IC~50~ | Monitors displacement of ATP site-binding fluorescent probes for binding assessment [102]. |
| KINOMEscan (DiscoveRx) | DNA-Tagging & qPCR | Dissociation Constant (K~d~) | Reports binding affinity; enables screening of hundreds of kinases in a high-throughput format [104]. |
| PhosphoSens (AssayQuant) | Continuous Fluorescence | Real-time Kinase Activity; IC~50~; K~inact~/K~I~ | Provides real-time kinetic progress curves, enabling mechanism of inhibition studies and work with complex lysates [105]. |
Protocol 1: Biochemical Selectivity Profiling Using a Commercial Panel
This protocol outlines the process for profiling a compound using a commercial kinase profiling service, such as SelectScreen (Thermo Fisher Scientific) [102].
- Compound Preparation: Prepare a 10 mM stock solution of the kinase inhibitor in DMSO. Dilute the compound in the appropriate assay buffer to create intermediate stocks for testing at the desired final concentrations (e.g., 0.1 µM and 1 µM).
- Assay Selection: Choose between kinase activity assays (e.g., Z'-LYTE, Adapta) or binding assays (e.g., LanthaScreen) based on the required information. ATP concentrations are typically used at levels near the K~m~app for each kinase.
- Primary Screening: Test the inhibitor in duplicate against a panel of recombinant human kinases (e.g., 156 kinases). Incubate the compound with the kinase and its substrate/ATP. The mean percentage inhibition for each kinase is calculated.
- Concentration-Response Curves: For kinases showing significant inhibition in the primary screen, perform a 10-point inhibitor titration to determine the IC~50~ value.
- Data Analysis: Plot the percentage of control activity against the log of the inhibitor concentration. Fit the data using non-linear regression to a four-parameter sigmoidal inhibition model to calculate IC~50~ values [102].
Diagram 1: Biochemical Profiling Workflow
Cellular Target Engagement Profiling
Cellular profiling provides critical information on cell permeability, stability, and target engagement in a more physiologically relevant context. Activity-based protein profiling (ABPP) is a powerful chemical proteomics method for identifying kinase drug targets directly in cell lysates or live cells.
Protocol 2: Site-Specific Competitive ABPP for Kinase Inhibitor Profiling
This protocol, adapted from van Bergen et al. (2025), uses phosphonate affinity tags to profile inhibitor targets with amino acid specificity [106].
- Sample Preparation: Treat cells with the kinase inhibitor of interest (e.g., NVP-BHG712) at varying concentrations (e.g., 0.1 nM to 10 µM) for a predetermined time. Prepare vehicle (DMSO)-treated control cells in parallel.
- Cell Lysis and Probe Labeling: Lyse the cells. Incubate the lysates with a broad-spectrum, activity-based kinase probe (e.g., XO44) to covalently label the ATP-binding pockets of active kinases.
- Peptide Digestion: Digest the labeled proteome into peptides using two complementary enzymes, such as trypsin and pepsin, to maximize kinase active site coverage.
- Enrichment of Labeled Peptides: Use affinity chromatography (e.g., streptavidin beads if using a biotinylated probe) to highly efficiently enrich the probe-labeled peptides.
- LC-MS/MS Analysis and Data Processing: Analyze the enriched peptides by liquid chromatography-tandem mass spectrometry (LC-MS/MS). Process the raw data using search engines like MaxQuant to identify the labeled peptides and quantify inhibitor engagement based on the reduction in probe labeling intensity compared to the DMSO control. Fit the dose-response data to determine IC~50~ values for each engaged kinase [106].
Diagram 2: Cellular Target Engagement Workflow
The Scientist's Toolkit: Research Reagent Solutions
Successful selectivity profiling relies on a suite of specialized reagents and platforms.
Table 3: Essential Reagents and Platforms for Kinase Selectivity Profiling
| Tool Category | Example Product/Platform | Function and Application |
|---|---|---|
| Commercial Profiling Services | SelectScreen (Thermo Fisher) [102] | Provides standardized, high-quality biochemical activity and binding data across large kinase panels. |
| Commercial Profiling Services | KINOMEscan (DiscoveRx) [104] | Offers high-throughput binding affinity (K~d~) screening against a vast kinome set. |
| Commercial Profiling Services | KinSight Services (AssayQuant) [105] | Delivers kinetic insight into compound potency, selectivity, and mechanism of inhibition using continuous assays. |
| Activity-Based Probes | XO44 and other pan-kinase ABPs [106] | Covalently label active kinase ATP-binding pockets for chemical proteomics and target engagement studies. |
| Detection Reagents | Pro-Q Diamond Dye [107] | A fluorescent phosphosensor for ultrasensitive global detection and quantitation of phosphorylated proteins on arrays or gels. |
| Detection Reagents | Phospho-specific Antibodies [9] | Enable detection and quantification of specific phosphorylated proteins in techniques like Western blot, ELISA, and flow cytometry. |
| Assay Kits & Reagents | PhosphoSens Assay Kits [105] | Fluorescent sensor peptide substrates for continuous, real-time measurement of kinase activity in biochemical or lysate-based formats. |
Data Integration and Application in Drug Discovery
Integrating data from multiple profiling methods provides a holistic view of inhibitor selectivity. Computational predictions can guide the selection of experimental panels, while experimental results from biochemical and cellular assays validate and refine the in silico models [102] [104]. This integrated data is crucial for:
- Lead Optimization: Chemistry efforts can be guided to eliminate problematic off-target activities while maintaining potency against the primary target.
- Target Validation: Selective tool compounds can be used to deconvolute complex biology and validate new kinase targets [104].
- Understanding Polypharmacology: Profiling can reveal intentional multi-targeted activity, which may be beneficial for overcoming drug resistance or treating complex diseases [103].
- Safety Assessment: Early identification of off-targets associated with known toxicities can help prioritize safer drug candidates for clinical development [102].
A multi-tiered strategy combining computational prediction, broad biochemical profiling, and cellular target engagement studies is the most effective approach for comprehensive kinase inhibitor selectivity assessment. The protocols and metrics detailed in this application note provide a roadmap for researchers to rigorously characterize compound specificity, ultimately enabling the development of safer and more effective kinase-targeted therapies.
Kinases represent one of the most targeted protein families in drug discovery, with their inhibitors broadly classified as either ATP-competitive or allosteric based on their binding sites and mechanisms of action [108] [109]. Understanding this distinction is critical for developing effective therapeutic strategies, as these inhibitor classes exhibit fundamental differences in selectivity profiles, resistance mechanisms, and conformational impacts on their kinase targets [110] [111]. ATP-competitive inhibitors bind directly to the active, ATP-binding site of the kinase, while allosteric inhibitors bind to regulatory sites remote from the catalytic center, inducing conformational changes that impair kinase function [109] [111]. This application note provides detailed methodologies for distinguishing these mechanisms through biochemical and cellular approaches, framed within the context of kinase activity and phosphorylation detection research.
Fundamental Mechanisms and Structural Consequences
Molecular Mechanisms of Inhibition
ATP-competitive inhibitors directly compete with ATP for binding to the kinase's catalytic cleft. They typically recognize the active "PH-out" conformation of kinases like AKT, where the pleckstrin homology (PH) domain disengages from the kinase domain [110] [111]. These inhibitors often paradoxically stabilize phosphorylation at activation loop residues (T308 and S473 in AKT) by protecting the kinase from phosphatases while simultaneously blocking substrate phosphorylation [110].
Allosteric inhibitors bind to regulatory sites distinct from the ATP-binding pocket. In AKT, for instance, they preferentially bind the "PH-in" conformation, where the PH domain interacts with the kinase domain, maintaining the kinase in an autoinhibited state [110] [111]. This binding prevents phosphorylation and activation of AKT, resulting in decreased phosphorylation at both T308 and S473 [110].
Conformational Impacts Revealed by Structural Biology
Hydrogen-deuterium exchange mass spectrometry (HDX-MS) studies reveal that these inhibitor classes induce distinct conformational changes in kinase structure. In AKT1, allosteric inhibitors cause substantive conformational changes that restrict membrane binding [111]. Conversely, ATP-competitive inhibitors induce extensive allosteric conformational changes that alter the autoinhibitory interface between PH and kinase domains, leading to increased membrane binding capacity as the PH domain becomes more accessible [111].
Research on ERK2 has further elucidated how ATP-competitive inhibitors can exhibit conformation selection properties, preferentially stabilizing specific conformational states (designated 'R' and 'L') that normally undergo reversible exchange in the apoenzyme [112]. This conformational selection can propagate to distal regions surrounding the activation loop, potentially influencing interactions with substrates and effectors [112].
The following diagram illustrates the fundamental mechanistic differences between these inhibitor classes:
Figure 1: Fundamental mechanisms of kinase inhibitor classes. Allosteric inhibitors stabilize inactive conformations, while ATP-competitive inhibitors bind active conformations and directly compete with ATP.
Experimental Approaches for Mechanism Elucidation
Kinase Activity Assays
Multiple assay platforms enable discrimination of inhibitor mechanisms through direct measurement of kinase activity. The Kinase Mobility Shift Assay (KiMSA) provides a non-radioactive method to quantify kinase activity using fluorescent-labeled peptide substrates (e.g., Kemptide-FITC for PKA) [37]. Phosphorylation increases peptide negative charge, causing a mobility shift during agarose gel electrophoresis that separates phosphorylated from non-phosphorylated species [37]. This enables direct assessment of inhibitor effects on kinase activity.
FRET-based biosensors like Picchu-B enable real-time monitoring of kinase activity in vitro [99]. These single-polypeptide biosensors link phospho-binding domains and kinase substrate peptides with flanking FRET pairs. Phosphorylation-induced conformational changes alter FRET efficiency, allowing kinetic analysis of inhibitor effects on EGFR and other kinases [99].
Protocol 1: KiMSA for Inhibitor Profiling
Reaction Setup: Prepare kinase reaction buffer (50 mM Tris-HCl pH 7.5, 10 mM MgCl₂, 1 mM DTT) containing ATP (variable concentrations for competition studies), fluorescent-labeled substrate peptide (e.g., Kemptide-FITC), and test inhibitors at desired concentrations [37].
Kinase Addition: Initiate reactions by adding purified kinase or cell lysates containing the kinase of interest. Include controls without inhibitor and without kinase.
Incubation: Conduct reactions for 25 minutes at 37°C in the dark to prevent fluorophore quenching [37].
Reaction Termination: Place samples on ice, add Tween-20 to 0.1%, and incubate at 100°C for 1 minute [37].
Electrophoresis: Resolve reaction products on 1.5-2% agarose gels in Tris-borate-EDTA buffer at 100V for 30-45 minutes.
Visualization & Quantification: Image gels using appropriate fluorescence detection. Calculate phosphorylation percentage from band intensities: Phosphorylation % = [Phosphopeptide/(Phosphopeptide + Non-phosphopeptide)] × 100 [37].
Phosphoproteomic Analysis and Kinase Activity Inference
Advanced phosphoproteomics enables system-wide assessment of inhibitor effects through kinase activity inference. The benchmarKIN package provides a comprehensive framework for evaluating kinase activities from phosphoproteomic data using kinase-substrate libraries [10]. This approach identifies deregulated kinases in biological contexts by analyzing phosphorylation patterns of known kinase targets [10].
Protocol 2: Phosphoproteomic Profiling for Inhibitor Mechanism Studies
Sample Preparation: Treat cells with ATP-competitive inhibitors, allosteric inhibitors, or vehicle control. Extract proteins under denaturing conditions with phosphatase inhibitors.
Phosphopeptide Enrichment: Digest proteins with trypsin, then enrich phosphopeptides using TiO₂ or IMAC chromatography [10].
LC-MS/MS Analysis: Analyze phosphopeptides by liquid chromatography coupled to tandem mass spectrometry.
Data Processing: Identify and quantify phosphorylation sites using computational tools like MaxQuant.
Kinase Activity Inference: Apply inference methods (KSEA, PTM-SEA, or ViPER) using kinase-substrate relationship databases (PhosphoSitePlus, SIGNOR) through the benchmarKIN platform [10].
Mechanism Discrimination: ATP-competitive inhibitors typically show broader effects on multiple kinases due to conserved ATP-binding pockets, while allosteric inhibitors demonstrate greater specificity with focused effects on targeted kinases and immediate downstream substrates [10] [109].
Cellular Signaling Analysis by Immunoblotting
Immunoblotting phosphorylation states of kinases and their substrates provides critical insights into inhibitor mechanisms. The distinct effects of ATP-competitive versus allosteric inhibitors on pathway biomarkers enable clear mechanistic discrimination.
Protocol 3: Signaling Pathway Analysis by Immunoblotting
Cell Treatment: Treat cells with inhibitors across a concentration range (e.g., 0.1-10 μM) and time course (0-24 hours). Include positive controls (growth factors, cytokines) for pathway activation.
Protein Extraction: Lyse cells in RIPA buffer containing protease and phosphatase inhibitors.
Immunoblotting: Separate proteins by SDS-PAGE, transfer to membranes, and probe with phosphospecific antibodies targeting:
- Activation loop phosphorylation of the target kinase (e.g., AKT T308/S473)
- Direct kinase substrates (e.g., PRAS40, GSK-3β for AKT)
- Downstream pathway effectors (e.g., S6, 4EBP1 for mTORC1 signaling) [110]
Mechanism Interpretation:
Table 1: Differential Signaling Effects of AKT Inhibitor Classes
| Parameter | Allosteric Inhibitors | ATP-competitive Inhibitors |
|---|---|---|
| AKT phosphorylation (T308/S473) | Decreased | Sustained or increased |
| Direct substrate phosphorylation (PRAS40, GSK-3β) | Decreased | Decreased |
| Downstream signaling (S6, 4EBP1) | Decreased | Variable (may be maintained through compensatory pathways) |
| Membrane binding | Restricted | Enhanced |
| Conformational state preference | PH-in (inactive) | PH-out (active) |
Resistance Mechanisms and Therapeutic Implications
Distinct resistance patterns emerge between inhibitor classes, informing therapeutic sequencing and combination strategies. Studies in prostate cancer models reveal that resistance to the allosteric AKT inhibitor MK-2206 frequently involves alterations in AKT isoforms themselves, such as AKT3 upregulation [110]. In contrast, resistance to the ATP-competitive inhibitor ipatasertib typically involves rewiring of compensatory pathways including PIM signaling, with AKT signaling to its direct substrates remaining impaired [110].
Critically, these resistance mechanisms demonstrate cross-resistance profiles: MK-2206-resistant cells maintain sensitivity to ipatasertib, while ipatasertib-resistant cells typically display cross-resistance to MK-2206 [110]. This differential resistance pattern provides a therapeutic strategy where allosteric inhibitor resistance can be overcome by switching to ATP-competitive inhibitors [110].
The following diagram illustrates the experimental workflow for distinguishing inhibitor mechanisms:
Figure 2: Integrated experimental workflow for distinguishing kinase inhibitor mechanisms. Multiple orthogonal approaches provide complementary data for definitive mechanistic classification.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Research Reagents for Kinase Inhibitor Mechanism Studies
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Fluorescent Kinase Substrates | Kemptide-FITC [37] | Fluorescent-labeled peptide substrate for kinase activity assays (e.g., KiMSA) |
| FRET-based Biosensors | Picchu-B [99] | Recombinant biosensors for real-time kinase activity monitoring in vitro |
| Allosteric Kinase Inhibitors | MK-2206 (AKT) [110] | Reference allosteric inhibitors for control experiments |
| ATP-competitive Kinase Inhibitors | Ipatasertib, AZD5363 (AKT) [108] [110] | Reference ATP-competitive inhibitors for control experiments |
| Phosphospecific Antibodies | Anti-pAKT (T308/S473), anti-pPRAS40 [110] | Immunoblot detection of pathway modulation |
| Kinase-Substrate Databases | PhosphoSitePlus, SIGNOR [10] | Curated kinase-substrate relationships for activity inference |
| Activity Inference Tools | benchmarKIN R package [10] | Computational analysis of kinase activities from phosphoproteomics |
Distinguishing between ATP-competitive and allosteric kinase inhibitor mechanisms requires integrated experimental approaches spanning biochemical, cellular, and system-wide levels. Orthogonal methodologies including kinase activity assays, signaling pathway analysis, and phosphoproteomic profiling provide complementary evidence for definitive mechanistic classification. The distinct conformational impacts, signaling modulation patterns, and resistance mechanisms between these inhibitor classes carry significant implications for therapeutic development, including inhibitor sequencing strategies and rational combination therapies to overcome resistance. As kinase inhibitor research advances, these mechanistic studies will continue to guide the optimal implementation of targeted kinase therapies in precision medicine.
Best Practices for Data Interpretation and Reporting in Publications and Regulatory Submissions
In the field of kinase research, the accurate interpretation and robust reporting of data from biochemical assays are foundational to drug discovery and development. Kinases regulate crucial intracellular pathways, and their dysregulation is linked to numerous diseases, including cancer, metabolic conditions, and neurodegenerative disorders [1]. The reliability of data emanating from kinase activity and phosphorylation detection assays directly impacts the validity of scientific publications and the success of regulatory submissions for new therapeutic agents. This application note details best practices for experimental protocols, data analysis, and reporting, framed within the rigorous context of biochemical kinase assays. Adherence to these practices ensures data integrity, facilitates peer review, and meets the stringent standards of regulatory bodies like the US Food and Drug Administration (FDA) and the Japan Pharmaceuticals and Medical Devices Agency (PMDA) [113].
Core Principles of Kinase Activity Assays
Fundamental Assay Types and Their Applications
Biochemical kinase assays are broadly categorized into activity assays and binding assays. The choice of assay format depends on the research question, desired sensitivity, throughput, and assay environment [1].
- Activity Assays: These directly measure the catalytic function of kinases by quantifying the formation of phosphorylated products or consequent changes in reaction components.
- Binding Assays: These assess the binding affinity of small molecules (e.g., inhibitors) to the kinase, often to the ATP-binding site or allosteric sites, without directly measuring catalytic output [1].
Key Technological Platforms
A variety of technologies are available for detecting kinase activity, each with distinct advantages. The table below summarizes the primary non-radioactive methods dominant in modern laboratories due to their scalability and safety [1] [9].
Table 1: Key Technology Platforms for Kinase Activity Detection
| Technology Platform | Detection Principle | Typical Readout | Key Applications |
|---|---|---|---|
| Luminescence-based | Quantifies ATP consumption or ADP formation [1]. | Luminescence | Universal kinase activity, high-throughput screening (HTS) [114]. |
| FRET/TR-FRET | Measures fluorescence resonance energy transfer between donor and acceptor molecules upon phosphorylation-induced proximity [99]. | Fluorescence ratio | Phosphorylation detection, real-time kinetic studies [1]. |
| Fluorescence Polarization (FP) | Detects change in rotational mobility of a fluorescent tracer upon binding to a kinase or being incorporated into a phosphorylated product [1]. | Polarization (mP units) | Binding affinity, competitive displacement assays [114]. |
| Fluorescence Quench/Superquenching | Uses fluorescent polymers and phosphate-binding interactions; phosphorylation inhibits quenching, generating a "turn on" signal [115]. | Fluorescence intensity | Protein substrate phosphorylation, HTS [115]. |
| ELISA | Uses phospho-specific antibodies in an immunoassay format to quantify phosphorylated substrates [1] [9]. | Colorimetric, Chemiluminescent | Specific phospho-site quantification, lower throughput studies [9]. |
| Mobility Shift | Separates phosphorylated from non-phosphorylated substrates based on charge or size using capillary electrophoresis [1]. | Fluorescence intensity | Direct, quantitative readouts without radioactivity [1]. |
| Transcreener (Immunodetection) | Directly immunodetects ADP formation using anti-ADP antibodies in FP, TR-FRET, or FI formats [99] [114]. | FP, TR-FRET, FI | Universal kinase activity, broad substrate applicability [114]. |
Experimental Protocols for Robust Data Generation
Protocol: TR-FRET-Based Kinase Activity Assay
This protocol is adapted for a high-throughput screening environment to identify kinase inhibitors, using a generic peptide substrate.
1. Reagent Preparation
- Kinase Buffer: 20 mM HEPES (pH 7.4), 5 mM MgCl₂, 1 mM DTT, 0.01% Triton X-100.
- ATP Solution: Prepare a 1 mM stock in kinase buffer.
- Substrate Solution: Prepare a fluorescently labeled peptide substrate stock in kinase buffer.
- Test Compounds: Prepare compound dilutions in DMSO, ensuring the final DMSO concentration in the assay is ≤1% (v/v).
- Detection Mix: Prepare TR-FRET detection reagents containing an antibody specific to the phosphorylated product, conjugated to a donor (e.g., Europium cryptate), and an acceptor (e.g., XL665) in a lysis/detection buffer [1] [99].
2. Assay Procedure
- Plate Setup: In a low-volume 384-well assay plate, add 2 µL of test compound or DMSO control.
- Enzyme/Substrate Addition: Add 4 µL of a pre-mixed solution containing the kinase and peptide substrate.
- Reaction Initiation: Initiate the reaction by adding 4 µL of ATP solution. The final reaction volume is 10 µL.
- Incubation: Seal the plate and incubate at room temperature for 60 minutes.
- Reaction Termination & Detection: Add 10 µL of the prepared TR-FRET detection mix to stop the reaction. Incubate for 30 minutes to allow signal development.
- Signal Readout: Read the plate on a compatible microplate reader capable of time-resolved FRET measurements (e.g., excitation at 337 nm, emission at 620 nm and 665 nm). The phosphorylation signal is proportional to the ratio of acceptor (665 nm) to donor (620 nm) emission [1].
3. Data Analysis
- Calculate the % inhibition for each compound using the formula:
% Inhibition = [1 - (Ratio_compound - Ratio_min_control) / (Ratio_max_control - Ratio_min_control)] * 100whereRatio_min_controlis the signal with no enzyme andRatio_max_controlis the signal with enzyme and DMSO. - Generate dose-response curves and calculate IC₅₀ values using non-linear regression analysis in appropriate software (e.g., GraphPad Prism).
Protocol: QTL Lightspeed Assay for Protein Substrate Phosphorylation
This protocol is designed for measuring kinase activity using natural, unmodified protein substrates, which can reveal inhibitors that bind to docking sites or induce unique conformational states [115].
1. Reagent Preparation
- Assay Buffer: As specified for the kinase (e.g., for PKCα: 20 mM HEPES pH 7.4, 5 mM MgCl₂, 0.1 mM CaCl₂, and relevant lipids) [115].
- Protein Substrate: e.g., 0.5 µg of Myelin Basic Protein (MBP) per reaction.
- QTL Lightspeed Beads: Fluorescent polymer-coated microspheres with metal-ion coordinating groups for phosphate binding [115].
- Tracer Peptide: A rhodamine-labeled phosphopeptide.
2. Assay Procedure
- Kinase Reaction: In a 384-well white Optiplate, perform the phosphorylation reaction in a 15 µL total volume containing the kinase, protein substrate, and ATP. Incubate for 1 hour at room temperature.
- Detection Step: Add the QTL Lightspeed beads and tracer peptide. The phosphorylated protein binds to the phosphate-binding sites on the beads, inhibiting the tracer peptide from associating. This prevents quenching of the fluorescent polymer, resulting in a "turn-on" signal.
- Signal Readout: Read the fluorescence intensity on a microplate reader at the appropriate wavelengths (e.g., excitation ~490 nm, emission ~520 nm) [115].
3. Data Analysis
- The increase in fluorescence correlates with the degree of protein substrate phosphorylation.
- Calculate Z'-factor to validate assay robustness:
Z' = 1 - [3*(σ_max_control + σ_min_control) / |μ_max_control - μ_min_control|]. A Z' > 0.5 is considered excellent for HTS [115]. - Determine IC₅₀ values for inhibitors as described in Section 3.1.
Data Interpretation, Validation, and Pathway Mapping
Statistical Analysis and Assay Validation
Robust data interpretation requires rigorous statistical validation.
Table 2: Key Statistical Parameters for Assay Validation
| Parameter | Definition | Acceptance Criterion | Application in Kinase Assays |
|---|---|---|---|
| Z'-Factor | A statistical parameter reflecting the assay signal dynamic range and data variation [115]. | Z' > 0.5 [115]. | Validates assay robustness for high-throughput screening campaigns. |
| Signal-to-Noise (S/N) | Ratio of the assay signal to the background noise. | S/N > 10 is desirable for a robust assay [115]. | Ensures the phosphorylation signal is sufficiently distinct from background. |
| Signal-to-Background (S/B) | Ratio of the mean signal of the positive control to the mean signal of the negative control. | Dependent on assay technology, but a higher ratio is preferred. | Used to assess the assay window. |
| Coefficient of Variation (%CV) | The ratio of the standard deviation to the mean, expressed as a percentage. | Typically < 10% for well-controlled assays. | Measures the precision and reproducibility of replicate samples. |
Troubleshooting and Mitigating Artefacts
Several common pitfalls can compromise data quality and interpretation [1]:
- Compound Interference: Fluorescent or colored compounds can quench signals or cause auto-fluorescence, leading to false positives/negatives. Countermeasures include using red-shifted fluorophores or label-free detection methods like the Transcreener platform [1] [114].
- Non-specific Inhibition: Molecules may inhibit kinases indirectly by chelating essential metal ions (e.g., Mg²⁺) or by forming aggregates. Cross-validation with an orthogonal assay format is recommended [1].
- Reagent Purity and Stability: Impurities in ATP, substrates, or buffers can significantly affect reaction kinetics. Use high-purity reagents and validate their stability under assay conditions.
- Enzyme Concentration: Use the minimal amount of kinase required to generate a robust signal to avoid substrate depletion and maintain linear initial velocity conditions.
Visualizing Signaling Pathways and Workflows
Understanding the kinase's position in a signaling network is crucial for interpreting inhibitory data in a physiological context. The diagram below illustrates a generic kinase signaling cascade leading to a cellular response.
Kinase Signaling Cascade
The experimental workflow for a typical kinase inhibition study is outlined below.
Kinase Assay Workflow
Reporting for Publications and Regulatory Submissions
Essential Elements for Scientific Publications
When publishing kinase data, provide sufficient detail to ensure reproducibility:
- Experimental Conditions: Explicitly state kinase concentration, substrate identity and concentration (peptide sequence if applicable), ATP concentration, buffer composition (pH, salts, detergents), and incubation time/temperature.
- Validation Data: Report key statistical parameters such as Z'-factor, S/N, and intra-assay %CV to demonstrate assay robustness [115].
- Control Data: Include full dose-response curves for standard inhibitors (e.g., Staurosporine) with reported IC₅₀ values to benchmark the assay performance.
- Data Representation: Clearly describe the number of biological and technical replicates, the error bars used (e.g., SD, SEM), and the statistical tests applied for significance.
Best Practices for Regulatory Data Integrity (ALCOA+)
Regulatory submissions require data that adheres to the ALCOA+ principles, ensuring data is Attributable, Legible, Contemporaneous, Original, Accurate, Complete, Consistent, Enduring, and Available [116]. This is critical for submissions to the FDA's Electronic Submissions Gateway (ESG) [116].
- Attributable & Contemporaneous: Maintain electronic audit trails that record who created or changed data and when. Do not back-date entries [116].
- Original & Accurate: Preserve raw data files from plate readers. Any data transformation should be documented and traceable. Validate submission software and processes [117] [116].
- Complete & Enduring: Ensure all data, including failed experiments, is retained for the required period. Keep thorough backups of submission packages and their associated metadata [116].
- Leverage Standards: Adhere to data standards such as those from the Clinical Data Interchange Standards Consortium (CDISC) for study data tabulation (SDTM) and analysis (ADaM) to facilitate regulatory review [113].
- Test Transmissions: Before live submission, use the FDA ESG test gateway to validate data transmission, encryption, and acknowledgment receipts (MDNs) [116].
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for Kinase Assays
| Reagent / Material | Function | Example / Specification |
|---|---|---|
| Recombinant Kinase | The enzyme target of the study. | Purified, full-length or catalytic domain, with verified activity. |
| Peptide/Protein Substrate | Phosphate acceptor in the kinase reaction. | Synthetic peptides (e.g., for Src: IYGEFKKK) or natural proteins (e.g., MBP, Histone H1) [115]. |
| ATP | Phosphate group donor. | High-purity, prepared fresh in buffer to avoid degradation. |
| Detection Kit | Reagents for quantifying phosphorylation or ADP formation. | TR-FRET, Transcreener FP, ADP-Glo [1] [114]. |
| Reference Inhibitor | Tool compound for assay validation and benchmarking. | Staurosporine (non-specific), or target-specific inhibitors with known potency [115]. |
| Multi-Mode Microplate Reader | Instrument for detecting assay signals. | Capable of reading luminescence, fluorescence, FRET, and polarization (e.g., PHERAstar FSX, CLARIOstar Plus) [79]. |
| Low-Volume Microplates | Reaction vessel for assays. | 384-well or 1536-well white, solid-bottom plates for optimal signal detection. |
| Data Analysis Software | For curve fitting and statistical analysis. | Software with non-linear regression for IC₅₀/EC₅₀ determination (e.g., GraphPad Prism). |
Conclusion
Biochemical kinase assays remain indispensable tools for unraveling kinase function and advancing therapeutic discovery. The field has evolved from traditional radioactive methods to sophisticated non-radioactive platforms and innovative biosensors that offer unprecedented sensitivity and live-cell capability. Successful implementation requires careful method selection based on specific research goals, rigorous optimization to minimize artifacts, and thorough validation to ensure data reliability. Future directions will focus on illuminating understudied kinases, developing more physiologically relevant assay systems, and creating advanced tools for real-time monitoring of kinase activity in complex biological environments. These advancements will continue to drive both fundamental understanding of cellular signaling and the development of next-generation kinase-targeted therapies for cancer, inflammatory diseases, and neurological disorders.
References
- 1. Biochemical assays for kinase activity detection [https://www.celtarys.com/science-highlights/biochemical-assays-kinase-activity.html]
- 2. Current technologies to identify protein kinase substrates in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4126243/]
- 3. Understanding and exploiting substrate recognition by ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2366800/]
- 4. Analysis of phosphorylation site occupancy of peptides ... [https://pubs.rsc.org/en/content/articlelanding/2025/ay/d4ay02208d]
- 5. Proteomic sensors for quantitative multiplexed and spatial ... [https://www.nature.com/articles/s41467-025-65950-2]
- 6. In vitro protein kinase activity measurement by flow cytometry [https://www.sciencedirect.com/science/article/abs/pii/S0003269708005812]
- 7. Methods for Detecting Phosphorylated Proteins and ... [https://pubmed.ncbi.nlm.nih.gov/39614054/]
- 8. Global comparative structural analysis of responses to ... [https://www.nature.com/articles/s41467-025-64116-4]
- 9. Methods for Detecting Protein Phosphorylation [https://www.rndsystems.com/resources/articles/methods-detecting-protein-phosphorylation]
- 10. Comprehensive evaluation of phosphoproteomic-based ... [https://www.nature.com/articles/s41467-025-59779-y]
- 11. Magnesium induced structural reorganization in the active ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11313852/]
- 12. Price To Be Paid for Two-Metal Catalysis: Magnesium Ions ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3446636/]
- 13. Kinase Screening Assay Services [https://www.reactionbiology.com/services/kinase-assays/kinase-screening/]
- 14. Protocol for detecting SnRK2 kinase activity in plants by ... [https://www.sciencedirect.com/science/article/pii/S2666166725002485]
- 15. Molecular docking and dynamics in protein serine ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1696204/full]
- 16. Dual-specificity mitogen-activated protein kinase ... [https://www.sciencedirect.com/science/article/pii/S0021925825024299]
- 17. Kinase activity of DYRK family members is required for ... [https://www.nature.com/articles/s42003-025-08373-5]
- 18. An atlas of bacterial serine-threonine kinases reveals ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12233097/]
- 19. Systematic Evaluation of Tyrosine Kinase Inhibitors as ... - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11417675/]
- 20. Dual-specific autophosphorylation of kinase IKK2 enables ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12208667/]
- 21. Analytical methods for protein kinase and inhibitor ... [https://www.sciencedirect.com/science/article/abs/pii/S0165993624005089]
- 22. Identification of Critical Phosphorylation Sites Enhancing ... [https://pubmed.ncbi.nlm.nih.gov/39617062/]
- 23. Dual-specific autophosphorylation of kinase IKK2 enables ... [https://elifesciences.org/articles/98009]
- 24. Dual-specific autophosphorylation of kinase IKK2 enables ... [https://elifesciences.org/reviewed-preprints/98009v2]
- 25. A critical evaluation of protein kinase regulation by activation loop autophosphorylation - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10359097/]
- 26. Exploring the conformational landscapes of protein kinases [https://pmc.ncbi.nlm.nih.gov/articles/PMC11346445/]
- 27. Structural Basis for the Regulation of Protein Kinase A by Activation Loop Phosphorylation - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3340281/]
- 28. Kinase Activation by Small Conformational Changes - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7899239/]
- 29. Assay Development for Protein Kinase Enzymes - NCBI - NIH [https://www.ncbi.nlm.nih.gov/books/NBK91991/]
- 30. Activation loop phosphorylation tunes conformational dynamics underlying Pyk2 tyrosine kinase activation - ScienceDirect [https://www.sciencedirect.com/science/article/abs/pii/S0969212623000370]
- 31. Kinase signaling cascades: an updated mechanistic landscape [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d5sc04657b?page=search]
- 32. Orally effective FDA-approved protein kinase targeted ... [https://www.sciencedirect.com/science/article/pii/S1043661825002300]
- 33. Non-Antibody Universal Detection of Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4363923/]
- 34. Molecular recording of cellular protein kinase activity with ... [https://www.nature.com/articles/s41589-025-01949-6]
- 35. ADP-Glo™ Kinase Assay Protocol [https://worldwide.promega.com/resources/protocols/technical-manuals/0/adp-glo-kinase-assay-protocol/]
- 36. Application of the [γ-32P] ATP kinase assay to study ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4116398/]
- 37. Kinase Mobility Shift Assay (KiMSA) for Assessing Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12245630/]
- 38. Application of scintillation proximity assay in drug discovery [https://pubmed.ncbi.nlm.nih.gov/16392890/]
- 39. Radiochemicals [https://www.revvity.com/category/radiochemicals]
- 40. Enzyme Assays: The Foundation of Modern Drug Discovery [https://bellbrooklabs.com/enzyme-assays-and-drug-discovery/?srsltid=AfmBOor2VNxrAGR9prBJTi-W4MiLwHIzeZpogR1GGyUULKXMLdGEJOHP]
- 41. Using Universal Inhibitor Screening Assays to Accelerate ... [https://bellbrooklabs.com/using-universal-inhibitor-screening-assays/?srsltid=AfmBOop2bk3AqvEfOENPoWQlxJwreOAJ6nyG49FxzWhBMy6buu7yDvQ5]
- 42. Spotlight: Activity-Based Kinase Assay Formats [https://www.reactionbiology.com/resources/reading-room/blog/spotlight-activity-based-kinase-assay-formats/]
- 43. Digital Cascade Assays for ADP- or ATP-Producing Enzymes ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10521141/]
- 44. Enzyme-coupled assays for simultaneous detection of ... [https://www.sciencedirect.com/science/article/pii/S016748891200225X]
- 45. Optimization of a Western blot protocol for the detection ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12455089/]
- 46. FastScan ™ Phospho-Akt (Ser473) ELISA Kit #80895 [https://www.cellsignal.com/products/elisa-kits/fastscan-phospho-akt-ser473-elisa-kit/80895?srsltid=AfmBOor0kewYaPZKawkjG3th2sI4qnH96pVHsDMoPhSQDRX4Ng8vY6VY]
- 47. Matched Antibody Pairs - ELISA [https://www.cellsignal.com/applications/elisa/matched-antibody-pairs?srsltid=AfmBOopwrOqgn6Tt-ZwOLb-eC_seLNl1O-NR4MaTMvXqoS8YDoQvbzvn]
- 48. An Introduction to Time-Resolved Fluorescence ... [https://columbiabiosciences.com/intro-to-tr-fret/]
- 49. LanthaScreen TR-FRET tyrosine kinase and protein ... [https://www.bmglabtech.com/en/application-notes/lanthascreen-tr-fret-tyrosine-kinase-and-protein-kinase-c-assay/]
- 50. Development of a direct high-throughput protein quantification ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9391279/]
- 51. Time-resolved Fluorescence Resonance Energy Transfer ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3064190/]
- 52. Macromolecular High‐Affinity Binding Probed by Advanced ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12376249/]
- 53. Fluorescent thermal shift-based method for detection of NF- ... [https://www.nature.com/articles/s41598-021-81743-1]
- 54. A general assay platform to study protein pharmacology ... [https://www.nature.com/articles/s41467-025-59658-6]
- 55. Application of Fluorescence- and Bioluminescence-Based ... [https://www.mdpi.com/2079-6374/14/12/570]
- 56. Genetically encoded fluorescent reporter for polyamines [https://pubmed.ncbi.nlm.nih.gov/40425580/]
- 57. Advances in quantitative high-throughput phosphoproteomics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8209658/]
- 58. Innovative strategies for measuring kinase activity to ... [https://www.sciencedirect.com/science/article/pii/S1359644624000321]
- 59. Utilization of Oriented Peptide Libraries to Identify ... [https://www.sciencedirect.com/science/article/pii/S002192581966120X]
- 60. Proteome-derived Peptide Libraries to Study the Substrate ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3734572/]
- 61. Methods for Detecting Protein Phosphorylation [https://www.bio-techne.com/resources/scientific-articles/methods-for-detecting-protein-phosphorylation]
- 62. Quenching (Fluorescence) - an overview [https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/quenching-fluorescence]
- 63. A fluorescent assay for alkaline phosphatase activity based ... [https://www.sciencedirect.com/science/article/abs/pii/S0925400519313449]
- 64. Accurate correction method and algorithm of fluorescence ... [https://www.sciencedirect.com/science/article/abs/pii/S1386142522012951]
- 65. Quenching (fluorescence) [https://en.wikipedia.org/wiki/Quenching_(fluorescence)]
- 66. LanthaScreen® Kinase Activity Assays [https://www.thermofisher.com/us/en/home/references/protocols/drug-discovery/kinase-protocols/lanthascreen-kinase-assay-basic-training-module/lanthascreen-kinase-activity-assays.html]
- 67. Suborganelle Sensing of Mitochondrial cAMP-Dependent ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2862470/]
- 68. Biochemical Assay Development: Strategies to Speed Up ... [https://bellbrooklabs.com/accelerate-biochemical-assay-development/?srsltid=AfmBOorQYink2GydHYkbZFdL4t6etwdomZwfc_fkJuaCGa1bEU5YEePE]
- 69. A General Guide for the Optimization of Enzyme Assay ... [https://www.sciencedirect.com/science/article/pii/S247255522206779X]
- 70. Optimizing enzyme inhibition analysis: precise estimation ... [https://www.nature.com/articles/s41467-025-60468-z]
- 71. DMSO induces major morphological and physiological ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12360551/]
- 72. Dimethyl Sulfoxide Suppresses Fusarium graminearum ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12488374/]
- 73. Dimethyl Sulfoxide as a Biocompatible Extractant for ... [https://www.mdpi.com/2305-6304/13/12/1038]
- 74. Unveiling the Solvent Effect: DMSO Interaction with Human ... [https://www.mdpi.com/1420-3049/30/14/3030]
- 75. Protocol HotSpot Kinase Assay [https://www.reactionbiology.com/assay-protocol-hotspot/]
- 76. Rapid Identification of Protein Kinase Phosphorylation Site ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4784478/]
- 77. Interference and Artifacts in High-content Screening - NCBI - NIH [https://www.ncbi.nlm.nih.gov/books/NBK615088/]
- 78. Clawing back: broadening the notion of metal chelators in ... [https://www.sciencedirect.com/science/article/abs/pii/S1367593112001652]
- 79. Kinase assays [https://www.bmglabtech.com/en/blog/kinase-assays/]
- 80. Enzyme Activity Assays for Protein Kinases [https://pmc.ncbi.nlm.nih.gov/articles/PMC5599267/]
- 81. Unveiling orphan receptor-like kinases in plants: novel ... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2024.1372361/full]
- 82. Kinase domain autophosphorylation rewires the activity ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9177650/]
- 83. Kinase inhibition profiles as a tool to identify ... [https://www.nature.com/articles/s41467-020-15428-0]
- 84. Identification of Critical Phosphorylation Sites Enhancing ... [https://www.sciencedirect.com/science/article/pii/S1535947624001798]
- 85. Evolution of orphan and atypical histidine kinases ... [https://www.sciencedirect.com/science/article/abs/pii/S0141813024044404]
- 86. How Pure is Pure? Understanding Reagent Purity Grades [https://www.biopharminternational.com/view/how-pure-is-pure-understanding-reagent-purity-grades]
- 87. The Most Common Grades of Reagents and Chemicals [https://www.labmanager.com/the-most-common-grades-of-reagents-and-chemicals-2655]
- 88. Purity and Grade of Chemical Reagent [https://en.tjconcord.com/article/175.html]
- 89. The Z prime value (Z´) [https://www.bmglabtech.com/en/blog/the-z-prime-value/]
- 90. From S/B to Z′: A Better Way to Measure Assay Quality in ... [https://bellbrooklabs.com/z-prime-a-better-way-to-measure-assay-quality-in-hts/?srsltid=AfmBOopPJBd-ERXVJbVvDqEFxMsA2WaIuRcx_naK1avvVe7sO_GA7sW6]
- 91. Z-factor [https://en.wikipedia.org/wiki/Z-factor]
- 92. Z' Does Not Need to Be > 0.5 - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7509605/]
- 93. Coefficient of variation [https://en.wikipedia.org/wiki/Coefficient_of_variation]
- 94. Calculating Inter- and Intra-Assay Coefficients of Variability [https://salimetrics.com/calculating-inter-and-intra-assay-coefficients-of-variability/]
- 95. Accelerating Kinase Drug Discovery with Validated ... [https://www.discoverx.com/accelerating-kinase-drug-discovery-with-validated-kinase-activity-assay-kits]
- 96. Development of a HTRF® Kinase Assay for Determination ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2774622/]
- 97. What Is the Best Kinase Assay? [https://bellbrooklabs.com/what-is-the-best-kinase-assay/?srsltid=AfmBOorqJ8TzslWpbhxsyvHq35U81o-FY0Gz6ftGbB-joVYUjwa3kawG]
- 98. Drug Discovery Solutions Kinase Report: Market , Forecast... Trends [https://www.researchandmarkets.com/reports/6090746/kinase-drug-discovery-solutions-market-report]
- 99. Rapid kinase activity detection and kinetic analysis using a ... [https://www.sciencedirect.com/science/article/abs/pii/S0304416525000996]
- 100. Cell Activity Assay Service 2025-2033 Trends and ... [https://www.datainsightsmarket.com/reports/cell-activity-assay-service-1404118]
- 101. Establishing correlation between in vitro potency and in ... [https://www.nature.com/articles/s41541-025-01181-2]
- 102. Computational analysis of kinase inhibitor selectivity using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6330000/]
- 103. The use of novel selectivity metrics in kinase research - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5217660/]
- 104. Extending kinome coverage by analysis of kinase inhibitor ... [https://www.sciencedirect.com/science/article/abs/pii/S1359644615000203]
- 105. AssayQuant: Continuous Kinase & Phosphatase Assays for ... [https://www.assayquant.com/]
- 106. Site-Specific Competitive Kinase Inhibitor Target Profiling ... [https://pubmed.ncbi.nlm.nih.gov/39826875/]
- 107. Quantitative analysis of protein phosphorylation status and ... [https://pubmed.ncbi.nlm.nih.gov/12872225/]
- 108. Inhibitors in AKTion: ATP-competitive vs allosteric - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7329346/]
- 109. Systematic comparison of competitive and allosteric kinase ... [https://pubmed.ncbi.nlm.nih.gov/33540355/]
- 110. Distinct resistance mechanisms arise to allosteric vs. ATP- ... [https://www.nature.com/articles/s41467-022-29655-0]
- 111. ATP-competitive and allosteric inhibitors induce differential ... [https://pubmed.ncbi.nlm.nih.gov/36758543/]
- 112. Conformation selection by ATP-competitive inhibitors and ... [https://elifesciences.org/articles/91507]
- 113. Navigating regulatory landscapes: A guide to global ... [https://www.iconplc.com/insights/blog/2024/06/04/navigating-regulatory-landscapes-guide-global-submission-standards]
- 114. How Does a Biochemical Kinase Assay Work? [https://bellbrooklabs.com/how-does-a-biochemical-kinase-assay-work/?srsltid=AfmBOor2-Pkk5yoVPZGL74v2-NqNKDivHD3WzYQLaXWkLk90lDERTiAA]
- 115. High-throughput kinase assays with protein substrates using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1166542/]
- 116. Data Integrity in Regulatory Submissions: Best Practices for ... [https://aayutechnologies.com/blog/data-integrity-in-regulatory-submissions/]
- 117. 8 Best Practices for Regulatory Data Management and ... [https://a-teaminsight.com/blog/8-best-practices-for-regulatory-data-management-and-reporting-upcoming-webinar/]