Advanced Calcium Signaling Detection: From Molecular Tools to Live-Cell Applications in Biomedical Research
This article provides a comprehensive overview of modern methods for detecting calcium signaling in live cells, a cornerstone technique for studying cellular physiology, neural computation, and drug mechanisms.
Advanced Calcium Signaling Detection: From Molecular Tools to Live-Cell Applications in Biomedical Research
Abstract
This article provides a comprehensive overview of modern methods for detecting calcium signaling in live cells, a cornerstone technique for studying cellular physiology, neural computation, and drug mechanisms. We explore the foundational principles of calcium indicators, from classic synthetic dyes to the latest genetically encoded sensors like jGCaMP8 and NEMOer. The scope extends to practical methodological guidance for application in various cell types and tissues, strategies for troubleshooting and optimizing imaging experiments, and a rigorous framework for validating and comparing tool performance. Tailored for researchers and drug development professionals, this review synthesizes current technical capabilities to empower robust experimental design and accurate data interpretation in both basic and translational research.
The Calcium Signaling Landscape: Principles, Ions, and Indicator Evolution
Calcium ions (Ca²⁺) function as a universal intracellular messenger, governing processes from embryonic development to neural computation. This whitepaper examines the sophisticated mechanisms of Ca²⁺ signaling, focusing on advanced detection methodologies that enable researchers to decipher its spatiotemporal dynamics in live cells. We explore the molecular players governing Ca²⁺ fluxes, detail cutting-edge genetically encoded calcium indicators (GECIs), and present computational tools for quantitative analysis of Ca²⁺ signaling data. The integration of these advanced technologies provides unprecedented insights into how Ca²⁺ encodes information through amplitude, frequency, and localization of signals to regulate diverse physiological functions, offering new targets for therapeutic intervention in various disease pathways.
Calcium ions represent one of the most versatile and ancient intracellular signaling systems in eukaryotic cells. As a second messenger, Ca²⁺ regulates a diverse array of cellular functions, from cell division and differentiation to cell death [1]. The extracellular environment maintains Ca²⁺ concentrations approximately 20,000-fold higher than the resting cytoplasmic concentration, creating a steep electrochemical gradient that allows rapid signaling upon channel activation [1]. This compartmentalization enables Ca²⁺ to function as both an extracellular first messenger and an intracellular second messenger, with its concentration under tight neuronal and hormonal control [1].
The universality of calcium signaling stems from its ability to transmit information through signal-specific patterns rather than simple concentration changes. Cells decode these patterns—including oscillation frequency, amplitude, and subcellular localization—to activate appropriate downstream responses [2]. This complex encoding mechanism allows Ca²⁺ to participate in numerous physiological and pathological processes, including organogenesis, tumorigenesis, proliferation, migration, and apoptosis [3]. Recent technical advances in live-cell imaging, genetically encoded indicators, and computational analysis have revolutionized our understanding of how Ca²⁺ dynamics regulate everything from fertilization to neural computation, making it an indispensable focus of modern cell signaling research.
Molecular Mechanisms of Calcium Signaling
Intracellular calcium signals originate from two primary sources: influx across the plasma membrane and release from intracellular stores. The endoplasmic reticulum (ER) serves as the major intracellular Ca²⁺ reservoir, with release mediated through inositol 1,4,5-trisphosphate receptors (IP₃Rs) and ryanodine receptors (RyRs) [1]. Store-operated calcium entry (SOCE) represents a crucial mechanism linking ER Ca²⁺ depletion to plasma membrane channel activation, maintaining Ca²⁺ homeostasis and sustaining longer-term signals [4].
Table 1: Major Calcium Channel Classes and Their Functions
| Channel Type | Activation Mechanism | Subcellular Localization | Primary Functions |
|---|---|---|---|
| Voltage-Operated Channels (VOCs) | Membrane depolarization | Plasma membrane | Neural excitation, muscle contraction |
| Store-Operated Channels (SOCs) | ER calcium depletion | Plasma membrane | Capacitative calcium entry, sustained signaling |
| Receptor-Operated Channels | Ligand binding | Plasma membrane | Synaptic transmission, local signaling |
| IP₃ Receptors | IP₃ binding | ER membrane | Intracellular calcium release, oscillation generation |
| Ryanodine Receptors | Calcium-induced calcium release | ER membrane | Excitation-contraction coupling, neural plasticity |
| Two-Pore Channels | NAADP binding | Acidic organelles | Local calcium signaling, organelle communication |
Additional complexity arises from specialized channels such as two-pore channel 2 (TPC2), responsible for Ca²⁺ release from acidic organelles like lysosomes and endosomes [5]. This diversity of sources and channels enables cells to generate highly specific Ca²⁺ signatures in response to different stimuli, allowing appropriate physiological responses.
Calcium Signaling in Cellular Processes
Calcium's role as a ubiquitous messenger is exemplified by its participation in numerous cellular processes. During neural computation, Ca²⁺ transients triggered by action potentials enable information processing across neural networks with millisecond precision [6]. The kinetics of these signals are critical, as electrical signals propagate through neural circuits over timescales of milliseconds, requiring rapid detection methods to track meaningful activity [6].
Beyond excitable cells, Ca²⁺ regulates development and tissue homeostasis through pathways like the Hippo signaling network, which controls organ size and regeneration. Recent research has identified Ca²⁺ as an emerging intracellular messenger for Hippo pathway regulation, where it transduces mechanical cues to the core signaling machinery [5] [7]. In this process, rearrangement of the actin cytoskeleton through mechanisms like calcium-activated actin reset (CaAR) constructs actin filaments that provide scaffolds for launching Hippo pathway activators such as protein kinase C (PKC) beta II [5].
The interplay between Ca²⁺ and other signaling pathways creates complex regulatory networks. For instance, simultaneous detection of dynamic calcium signaling and ERK activity reveals bidirectional communication, where calcium signaling can regulate ERK activity through multiple mechanisms, and conversely, ERK signaling can affect the triggering and intensity of calcium signaling [3]. This complexity underscores the importance of multi-parameter live-cell imaging for comprehensive understanding of cellular signaling networks.
Detection Methods and Experimental Approaches
Genetically Encoded Calcium Indicators
Genetically encoded calcium indicators (GECIs), particularly the GCaMP series, have revolutionized calcium signaling research by enabling targeted expression in specific cell types and subcellular compartments. These protein-based indicators combine a calcium-binding protein (calmodulin) with a fluorescent protein, producing fluorescence changes upon calcium binding [6].
Recent engineering efforts have produced GCaMP variants with dramatically improved kinetics and sensitivity. The jGCaMP8 series represents the state-of-the-art, with three principal variants optimized for different applications: jGCaMP8s (sensitive) exhibits the highest signal-to-noise ratio for detecting single action potentials; jGCaMP8f (fast) has ultra-fast kinetics suitable for tracking high-frequency neural activity; and jGCaMP8m (medium) offers a balance between sensitivity and kinetics [6].
Table 2: Performance Characteristics of Advanced GECIs
| Indicator | 1AP ΔF/F₀ (%) | Half-Rise Time (ms) | Half-Decay Time (ms) | Primary Applications |
|---|---|---|---|---|
| jGCaMP8s | ~485 | 9.3 | 610 | Detecting single action potentials under optimal conditions |
| jGCaMP8m | ~330 | 6.9 | 240 | General-purpose imaging with balanced properties |
| jGCaMP8f | ~180 | 6.6 | 110 | Tracking neural populations with high temporal resolution |
| jGCaMP7f | ~175 | 22.2 | 480 | Previous generation fast variant |
| XCaMP-G | ~105 | 12.5 | 190 | Alternative design with different spectral properties |
These indicators enable detection of individual action potentials with millisecond precision, allowing researchers to track large populations of neurons on timescales relevant to neural computation [6]. The nearly tenfold faster fluorescence rise times of jGCaMP8 sensors compared to previous GCaMPs represent a quantum leap in our ability to capture rapid calcium dynamics in live cells and intact organisms.
Ratiometric Imaging and FRET-Based Sensors
Ratiometric imaging approaches provide internal calibration that minimizes artifacts from variations in indicator concentration, path length, or illumination intensity. Fura-2, a chemical indicator with dual excitation wavelengths, has been widely used for this approach [4]. For genetically encoded sensors, FRET (Förster Resonance Energy Transfer)-based indicators consisting of donor and acceptor fluorescent proteins linked by a calcium-sensitive element offer similar benefits [8].
A highly sensitive calcium FRET biosensor based on ECFP and YPet has been developed for visualizing intracellular Ca²⁺ signaling with high spatiotemporal resolution [8]. This biosensor enables quantification of calcium responses through ratio imaging, calculating IYPet/IECFP to generate quantitative measurements of calcium dynamics independent of sensor concentration [8]. Such FRET-based approaches are particularly valuable for experiments requiring precise quantification of absolute calcium concentrations or when monitoring long-term changes where artifacts might confound intensity-based measurements.
Experimental Workflow for Calcium Imaging
The experimental workflow for calcium signaling studies typically involves multiple stages from sample preparation to data analysis. For studies using genetically encoded indicators, the process begins with delivery of the indicator to target cells via transfection, viral transduction, or transgenic approaches. Cells are then mounted on an appropriate imaging system, often with environmental control to maintain physiological conditions during imaging.
Figure 1: Experimental workflow for calcium signaling studies, highlighting the integration of wet-lab and computational phases.
Following image acquisition, computational analysis extracts quantitative parameters from the calcium signals. Region-of-interest (ROI) detection identifies active cellular regions, followed by signal extraction and parameter quantification. This workflow enables researchers to transform raw imaging data into mechanistic understanding of calcium signaling events in live cells.
Quantitative Analysis of Calcium Signaling Data
Computational Tools and Approaches
The complexity and size of modern calcium imaging datasets necessitate sophisticated computational tools for quantitative analysis. Traditional off-the-shelf software often proves inadequate for the specific needs of specialized experiments, leading many research groups to develop custom analysis pipelines [4]. A collection of Jupyter-Lab "notebooks" implemented in Python has been developed specifically to address these challenges, providing flexibility for different experimental paradigms while ensuring quantifiable, consistent, and repeatable analysis [4].
These computational tools employ algorithms from computer graphics and image processing, including Canny edge detection for locating cell outlines, flood fill for identifying regions of interest, and watershed segmentation for separating overlapping cellular regions [4]. The notebooks are organized into two categories: main processing (ROI identification) and optional post-processing (peak identification, frequency analysis, and visualization). This modular approach allows researchers to apply appropriate analysis methods for different experimental designs, including single wavelength indicator experiments, agonist-induced Ca²⁺ signaling, store-operated calcium entry (SOCE), and inhibitor efficacy studies [4].
Key Parameters in Calcium Signal Analysis
Calcium signaling data analysis focuses on extracting specific parameters that characterize the temporal and spatial properties of calcium transients. For neural activity monitoring, these include latency (time from stimulus to response onset), rise time (signal progression to peak), decay time (return to baseline), amplitude (peak intensity change), frequency (oscillation rate), and spatial spread (propagation through cellular compartments) [4] [6].
Table 3: Quantitative Parameters for Calcium Signal Characterization
| Parameter | Definition | Biological Significance | Analysis Method |
|---|---|---|---|
| Latency | Time from stimulation to response onset | Signal transduction speed | Threshold crossing relative to stimulus |
| Rise Time | Time from onset to peak amplitude | Calcium release/diffusion kinetics | Time between 10% and 90% of peak amplitude |
| Decay Time | Time from peak to baseline recovery | Calcium clearance efficiency | Exponential fitting or return to baseline |
| Amplitude (ΔF/F₀) | Fractional fluorescence change | Strength of cellular response | (Fpeak - Fbaseline) / F_baseline |
| Frequency | Oscillation rate in periodic signals | Information encoding in frequency domain | Peak detection with temporal filtering |
| Area Under Curve | Integral of signal over time | Total calcium load | Numerical integration of ΔF/F₀ |
| Spatial Spread | Propagation through cellular regions | Intercellular communication | Analysis of signal correlation between ROIs |
Peak detection algorithms are particularly important for analyzing calcium transients, employing mathematically defined criteria to identify significant peaks while minimizing subjective bias [4]. These algorithms typically incorporate user-adjustable parameters for noise tolerance, minimum peak prominence, and minimum distance between peaks to accommodate different signal characteristics across experimental paradigms.
Frequency Analysis and Signal Propagation
For oscillatory calcium signals, frequency domain analysis provides insights into information encoding mechanisms. Calcium oscillations of varying frequency can activate different transcriptional programs, making frequency analysis crucial for understanding downstream effects [4]. The Frequency_Analysis notebook in the computational toolkit enables quantification of oscillation characteristics, including dominant frequencies, regularity, and phase relationships between different cellular regions [4].
Spatial analysis of calcium waves reveals how signals propagate through individual cells and cell populations. Tools for creating ratio images over time and analyzing signal correlation between adjacent ROIs help researchers understand intercellular calcium communication [8]. These analyses are particularly important in tissues where coordinated calcium signaling regulates processes such as secretion, contraction, and neural synchronization.
Advanced Applications and Integrated Sensing
Simultaneous Multi-Parameter Imaging
A cutting-edge advancement in calcium signaling research involves the simultaneous detection of multiple signaling activities in live cells. Recent protocols enable dynamic, synchronous recording of calcium signals and ERK activity in living cells using stable expression of multiple genetically-encoded probes and multi-channel confocal microscopy [3]. This approach addresses the spatiotemporal encoding dynamic mechanism of both signaling pathways and reveals their interactions and causal relationships.
The methodology utilizes spectrally distinct biosensors with minimal overlap in excitation and emission spectra, allowing simultaneous imaging without cross-talk. For example, combining green GCaMP sensors with red or near-infrared ERK biosensors enables researchers to monitor both signaling activities in real-time, revealing how calcium transients regulate MAPK signaling and vice versa [3]. Such integrated approaches are essential for understanding signaling networks rather than isolated pathways, providing more comprehensive models of cellular information processing.
Microenvironment and Calcium Signaling
The cellular microenvironment profoundly influences calcium signaling through mechanical forces, substrate stiffness, and spatial constraints. Studies using micro-patterned surfaces demonstrate that physical constraints can modulate calcium responses to stimuli such as ATP [8]. Human umbilical vein endothelial cells (HUVECs) cultured on different surface micro-patterns showed shorter decay times and reduced peaks of intracellular calcium transients compared to unconstrained cells [8].
Voltage-operated channels (VOCs) have been identified as key mediators linking microenvironment to calcium responses. When HUVECs were constrained on micro-patterns, inhibition of VOCs eliminated the effect of different patterns on calcium signals [8]. Furthermore, when two connected HUVECs were constrained to grow on a micro-pattern, they exhibited drastically distinct calcium responses upon ATP stimulation, in contrast to the similar responses of connected cells cultured without patterns [8]. This phenomenon was also VOC-dependent, highlighting how physical microenvironment shapes calcium signaling through specific channel regulation.
Figure 2: Signaling pathway showing how microenvironment regulates cellular responses through calcium-mediated mechanisms.
Calcium Signaling in Disease Pathways
Dysregulation of calcium signaling contributes to numerous pathological conditions, making it an important therapeutic target. In cancer biology, calcium channels and pumps are frequently altered, affecting processes such as proliferation, migration, and apoptosis [5] [7]. For example, in breast cancer cells, expression of secretory pathway Ca²⁺-ATPase 2 (SPCA2) inhibits epithelial-to-mesenchymal transition by increasing cellular Ca²⁺ levels and promoting E-cadherin expression, which subsequently activates the Hippo pathway through Lats1/2 kinase [7].
In neurodegenerative diseases, disrupted calcium homeostasis contributes to neuronal dysfunction and death. Mutations in various calcium channels have been linked to Alzheimer's disease, Parkinson's disease, and Huntington's disease, making the restoration of normal calcium signaling a promising therapeutic strategy. The development of more sensitive calcium imaging techniques enables researchers to track these dysregulations in disease models and evaluate potential interventions.
The Scientist's Toolkit: Essential Research Reagents
Table 4: Essential Research Reagents for Calcium Signaling Studies
| Reagent Category | Specific Examples | Key Functions | Applications |
|---|---|---|---|
| Genetically Encoded Calcium Indicators | jGCaMP8 series, XCaMP series, FRET-based biosensors | Visualizing calcium dynamics in specific cell types | Live-cell imaging, in vivo monitoring |
| Chemical Calcium Indicators | Fura-2, Indo-1, Fluo-4 | Ratiometric or intensity-based calcium detection | Cell population studies, high-throughput screening |
| Channel Modulators | Store-operated channel inhibitors, TRPV agonists/antagonists | Probing specific calcium entry pathways | Mechanistic studies, target validation |
| Calcium Pumps and Channel Antibodies | SERCA, PMCA, IP₃R antibodies | Localizing endogenous calcium handling proteins | Immunocytochemistry, Western blotting |
| Recombinant Adenoviral Vectors | Adeno-X Expression System | Efficient delivery of biosensors to primary cells | Studies in non-dividing or hard-to-transfect cells |
| Micro-patterning Reagents | Comb polymer solutions, PDMS molds | Controlling cellular microenvironment | Mechanobiology studies, single-cell analysis |
This toolkit enables researchers to design experiments addressing specific aspects of calcium signaling, from basic characterization of calcium transients to sophisticated manipulation of signaling pathways in physiologically relevant contexts. The selection of appropriate reagents depends on the experimental goals, cell type, and required temporal and spatial resolution.
Calcium signaling represents a fundamental regulatory system operating across biological scales, from fertilization to neural computation. The development of advanced detection methods, particularly genetically encoded calcium indicators with improved sensitivity and kinetics, has transformed our ability to monitor these signals in live cells and intact organisms. Coupled with sophisticated computational tools for quantitative analysis, researchers can now decipher the complex language of calcium signaling with unprecedented precision.
The integration of calcium imaging with other signaling biosensors and microenvironmental control approaches provides multidimensional insights into how cells process information through this ubiquitous messenger. These technical advances continue to drive discoveries in basic biology and disease mechanisms, offering new opportunities for therapeutic interventions targeting calcium signaling pathways in various disorders. As these methodologies evolve, they will undoubtedly reveal new dimensions of calcium's role as a master regulator of cellular function.
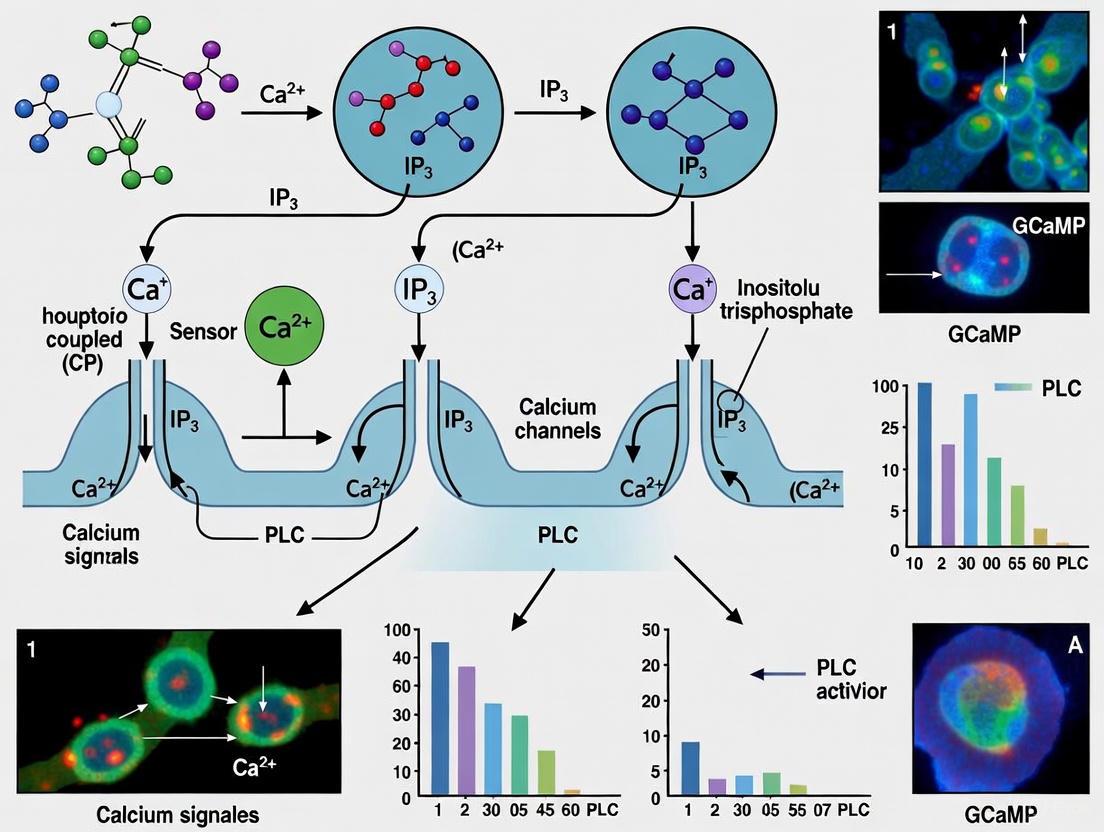
Calcium ions (Ca²⁺) are ubiquitous intracellular messengers that regulate a vast array of cellular processes, from neurotransmission and muscle contraction to gene expression and cell death. In neuroscience, the study of calcium dynamics has revolutionized our understanding of cellular communication, revealing intricate signaling patterns that operate across multiple spatial and temporal scales. The spatiotemporal organization of Ca²⁺ signals—including brief, localized spikes; propagating waves; and highly confined microdomains—encodes specific information that enables cells to perform complex computations and respond appropriately to stimuli. This technical guide examines the mechanisms, detection methodologies, and functional implications of these dynamic calcium signaling patterns, framed within the context of modern live-cell imaging research. Recent advances in imaging technologies, genetically encoded calcium indicators (GECIs), and computational analysis tools have provided unprecedented insights into how these coordinated signals shape brain function in health and disease, offering new avenues for therapeutic intervention in neurological disorders [9] [10].
Established Mechanisms of Intracellular Calcium Signaling
Intracellular calcium dynamics are governed by a complex interplay of release from internal stores and influx across the plasma membrane. The endoplasmic reticulum (ER) serves as a major calcium reservoir, with release primarily mediated by inositol 1,4,5-trisphosphate receptors (IP3Rs) and ryanodine receptors (RyRs). These receptors are activated by various second messengers and calcium itself, enabling complex regulatory dynamics [9] [11]. Additional mechanisms include store-operated calcium entry (SOCE) mediated by STIM and Orai proteins, voltage-gated calcium channels (VGCCs), and reverse operation of the Na⁺-Ca²⁺ exchanger (NCX) during elevated intracellular sodium [9]. The specific combination of these pathways varies by cell type, allowing for specialized calcium signaling profiles tailored to cellular function.
Table 1: Principal Sources of Calcium Elevations in Cells
| Mechanism/Source | Mode of Activation | Functional Output |
|---|---|---|
| IP3R | G-protein-coupled receptor activation → phospholipase C → IP3 production → ER Ca²⁺ release | Gliotransmitter release, metabolic coupling |
| SOCE | ER Ca²⁺ depletion → STIM sensor activation → Orai channel opening | ER Ca²⁺ store replenishment, sustained signaling |
| mGluR | Glutamate binding → phospholipase C activation → IP3-mediated Ca²⁺ mobilization | Gliotransmission, synaptic modulation |
| P2Y Purinergic Receptors | ATP/ADP binding → phospholipase C activation → IP3-mediated Ca²⁺ mobilization | Gliotransmission, intercellular Ca²⁺ wave propagation |
| VGCC | Membrane depolarization → channel opening → Ca²⁺ influx | Localized Ca²⁺ entry, depolarization-linked responses |
| NCX Reverse Mode | Elevated intracellular Na⁺ (e.g., after neurotransmitter uptake) | Ca²⁺ entry independent of ER stores, ionic homeostasis |
Spatial Organization of Calcium Signals
The spatial architecture of calcium signals ranges from highly localized microdomains to global waves that propagate through entire cellular networks. Microdomains represent nanoscale calcium elevations in subcellular compartments such as synaptic boutons or dendritic spines, where restricted geometry enables precise control of calcium-dependent processes without affecting distant cellular regions. These microdomains arise through the coordinated opening of clusters of calcium channels and are particularly important for regulating neurotransmitter release and synaptic plasticity [9]. At an intermediate scale, calcium waves propagate through larger cellular territories via intracellular diffusion or intercellular coupling. Gap junctions composed of connexins allow direct cell-to-cell movement of calcium and other messengers, while paracrine signaling through extracellular ATP enables propagation across non-directly connected cells [9] [11]. This hierarchical spatial organization enables cells to operate at both local and global scales, fine-tuning individual synaptic events while coordinating activity across broader networks.
Temporal Dynamics of Calcium Transients
Calcium signals exhibit diverse temporal patterns that encode specific information. Fast transients (milliseconds to seconds) often follow bursts of neuronal activity and regulate rapid processes like vesicle release. Slow oscillations (seconds to minutes) shape longer-term network states and can frequency-modulate neuronal excitability. The kinetics of these signals are determined by the interplay between calcium release/influx mechanisms and removal systems including pumps, exchangers, and buffers. Activity-dependent modulation ensures that calcium elevations scale with synaptic demand, creating a dynamic feedback system that adapts to changing circuit conditions [9]. In vivo imaging studies have revealed that sensory stimulation evokes robust calcium responses in cortical astrocytes that are temporally aligned with neuronal firing patterns and behavioral states, highlighting the tight coupling between calcium dynamics and functional output [9].
Calcium Wave Propagation: Modeling and Dynamics
Mechanisms of Wave Propagation
Traveling calcium waves represent a fundamental mode of long-distance signaling in both neurons and glia. These waves are mediated primarily by calcium-induced calcium release (CICR) through IP3Rs and RyRs, creating a regenerative process that propagates through the cytoplasm. Wave characteristics are highly dependent on the complement and density of these receptors. When both IP3Rs and RyRs are functional, wave speeds typically range from 100 to several hundred micrometers per second with cytosolic calcium transients reaching tens of micromolar. In contrast, when RyRs are absent, these values decrease to tens of micrometers per second and 1-6 micromolar, respectively [11]. The spatial distribution of receptors within cells is often non-uniform, with concentrated "hotspots" serving as initiation points or amplification sites for wave propagation. These areas are implicated in localized calcium "puffs" or "sparks" that can serve as wave nucleation points [11].
Modeling Wave Dynamics in Cellular Structures
Computational modeling has been instrumental in understanding calcium wave propagation. Recent modeling efforts have examined wave dynamics in idealized cellular morphologies including dendrite-like processes and cell bodies. These models incorporate representations of IP3Rs, RyRs, and other transport mechanisms, supporting fully regenerative traveling waves for significant parameter ranges [11]. Sensitivity analyses reveal that wave characteristics are most dependent on receptor areal densities and the diffusion coefficient for cytoplasmic calcium. Models have also identified Hopf bifurcations between stable and unstable regimes, with the latter characterized by periodic calcium spikes. Interestingly, traveling waves are possible in unstable processes during phases with sufficiently high calcium levels in the endoplasmic reticulum [11]. For some parameter values, damped and abortive waves are observed, suggesting mechanisms for wave termination.
Diagram: Calcium Wave Propagation Mechanism
Table 2: Calcium Wave Characteristics Under Different Receptor Conditions
| Parameter | IP3Rs + RyRs Functional | RyRs Absent |
|---|---|---|
| Wave Speed | 100 - several hundred µm/s | Tens of µm/s |
| Cytosolic [Ca²⁺] Amplitude | Tens of µM | 1 - 6 µM |
| Primary Mechanism | Calcium-induced calcium release | IP3 diffusion & binding |
| Spatial Range | Long-distance propagation | More localized spread |
| Stability | Periodic spiking possible | More graded responses |
Advanced Detection Methods for Calcium Dynamics
Fluorescent Indicators and Imaging Modalities
The study of calcium dynamics relies heavily on fluorescent indicators that transduce calcium concentration into measurable optical signals. These include chemical calcium-sensitive dyes (e.g., Oregon Green 488 BAPTA-1 AM) and genetically encoded calcium indicators (GECIs, e.g., GCaMP series). Chemical dyes offer simplicity of use and work across various species without genetic manipulation, but they lack cell-type specificity and can exhibit dye leakage over time [12]. GECIs provide targeted expression to specific cell types, enable long-term repeated imaging, and permit chronic studies in awake, behaving animals [12] [13]. Imaging modalities span from widefield microscopy for large-scale population imaging to two-photon microscopy for deeper tissue penetration with reduced scattering. Each approach presents tradeoffs between spatial coverage, temporal resolution, and depth penetration that must be balanced according to experimental needs [10].
Computational Analysis Tools and Pipelines
The complexity and scale of modern calcium imaging data require sophisticated computational tools for processing and analysis. Recent advances have produced several specialized software packages:
CalciumNetExploreR (CNER) is an R package that streamlines the analysis of time-series calcium imaging data through an integrated pipeline including normalization, binarization, network construction, and topological analysis. It calculates metrics such as clustering coefficients, global efficiency, and community structures to characterize functional connectivity [14] [15].
NeuroSpikeX provides comprehensive detection and characterization of neuronal calcium dynamics, offering robust spike detection and network metrics through a user-friendly interface that integrates with existing workflows [16].
CalTrig is a GUI-based tool that integrates machine learning for calcium transient identification in freely moving rodents. It combines manual, parameter-based, and machine learning approaches (GRU, LSTM, Transformer models) for flexible and accurate transient detection [13].
DeepWonder addresses the particular challenges of widefield calcium imaging through a deep-learning approach that effectively removes background contamination. This tool achieves nearly tenfold processing speed acceleration and improved neuronal extraction compared to conventional methods like CNMF-E [10].
AQuA2 (Activity Quantification and Analysis) is a versatile platform that decomposes complex live-imaging datasets into elementary signaling events using machine learning, enabling quantification of molecular activities and identification of consensus functional units across diverse biosensors and experimental conditions [17].
Diagram: Calcium Imaging Analysis Workflow
Experimental Protocols for Calcium Imaging
In Vivo Calcium Imaging in Freely Moving Rodents
Comprehensive protocol for studying calcium dynamics in awake, behaving animals:
Surgical Procedures:
- Virus Injection: Anesthetize mice with isoflurane (2.5% for induction, ~1.2% for maintenance). Inject AAV vectors encoding GECIs (e.g., AAV1-Syn-jGCaMP8f-WPRE) into target brain regions using stereotaxic coordinates. For medial prefrontal cortex (mPFC): AP +2.05 mm, ML ±0.3 mm, DV -2.45 mm from bregma. Use a 28-gauge injection needle to deliver 0.5 µl/site at 0.1 µl/min using a precision syringe pump. Leave the needle in place for 5 minutes post-injection to prevent backflow [13].
- Lens Implantation: Immediately after virus injection, lower a GRIN lens (diameter 1.0 mm, length ~4.0 mm, working distance 200 µm) through the cranial window to 200 µm above the injection site. Seal the space between the lens and skull with surgical silicone and secure with dental cement [13].
- Base Plating: Three weeks post-injection, mount a metal baseplate over the lens using super glue gel, guided by a MiniScope for optimal field of view. Attach a protective cap when not imaging [13].
Data Acquisition:
- Record calcium activity at 10-60 Hz frame rate with appropriate spatial resolution (0.8-1.0 µm pixel size) during behavioral tasks. Simultaneously track animal behavior using complementary recording systems [13].
Data Processing:
- Use Minian or similar pipelines for initial processing including motion correction, source extraction, and calcium trace generation.
- Apply CalTrig for post-processing evaluation, synchronized visualization, and calcium transient identification using integrated machine learning models [13].
Spinal Cord Calcium Imaging Protocol
Method for monitoring calcium dynamics in spinal cord circuits:
Surgical Preparation:
- Perform terminal exposure surgery to access the dorsal horn of the spinal cord. Alternatively, implant viewing chambers or microprisms for repeated measurements over days or weeks without anesthesia [12].
Indicator Loading:
- For non-specific labeling, use calcium-sensitive fluorescent dyes (e.g., Oregon Green 488 BAPTA-1 AM) loaded into dorsal horn cells after spinal transection [12].
- For cell-type-specific imaging, use transgenic expression or viral delivery (AAV-GCaMP6) under cell-type-specific promoters to target neurons or glia [12].
Imaging and Analysis:
- Image layers I-IV of the dorsal horn where somatosensory inputs are processed. These superficial layers are accessible for in vivo fluorescence microscopy.
- Monitor tens to hundreds of neurons or glial cells simultaneously to identify population-level activity patterns in response to sensory stimuli [12].
- Apply network analysis tools to decode functional connectivity and coordinated activity patterns.
Table 3: Research Reagent Solutions for Calcium Imaging Studies
| Resource | Type | Primary Function | Applications |
|---|---|---|---|
| GCaMP Series | Genetically Encoded Calcium Indicator | Fluorescent calcium sensing | Long-term in vivo imaging, cell-type-specific monitoring |
| Oregon Green 488 BAPTA-1 AM | Chemical calcium dye | Single-use calcium sensing | Acute preparations, multi-species studies |
| AAV Vectors | Viral delivery system | GECI expression in target cells | Specific neural circuit labeling |
| Minian | Computational software | Calcium trace extraction from video | Processing of one-photon miniscope data |
| Suite2p | Computational software | Calcium trace extraction | Processing of two-photon imaging data |
| CalciumNetExploreR | R package | Network analysis of calcium dynamics | Functional connectivity, topology studies |
| DeepWonder | Deep learning tool | Neuron extraction from widefield imaging | Large-scale population analysis |
| CalTrig | GUI-based tool | Calcium transient identification | Machine learning-based detection in behaving animals |
| AQuA2 | Analysis platform | Spatiotemporal signal quantification | Event-based analysis across diverse biosensors |
Functional Implications and Pathophysiological Correlates
The spatiotemporal dynamics of calcium signals have profound functional consequences for cellular computation and communication. In astrocytes, calcium elevations regulate gliotransmitter release (glutamate, ATP, GABA, D-serine), ion homeostasis, metabolic support, and morphological plasticity [9]. These processes enable bidirectional communication with neurons, influencing synaptic efficacy and network stability. The specific spatial and temporal patterns of calcium signals determine their functional impact: localized microdomains enable precise regulation of individual synapses, while global waves coordinate activity across broader neural circuits [9].
Disrupted calcium signaling is implicated in numerous pathological conditions. In epilepsy, aberrant astrocytic calcium signaling contributes to network instability through dysregulated gliotransmission and impaired ion regulation. Alzheimer's disease involves compromised calcium homeostasis that leads to synaptic dysfunction, while Parkinson's disease exhibits calcium dysregulation that contributes to neuronal vulnerability. Neurodevelopmental disorders also show impaired maturation of calcium signaling pathways [9]. Understanding these pathophysiological mechanisms provides potential avenues for therapeutic intervention targeting calcium signaling components.
Recent advances in detection methods have revealed that extracellular calcium itself functions as a dynamic signaling mediator, influencing neuronal excitability within milliseconds through mechanisms such as calcium-sensing receptor (CaSR) activation, ion channel modulation, and ephaptic coupling [9]. This expanded view of calcium signaling, encompassing both intracellular and extracellular dynamics, offers a more comprehensive framework for understanding neural computation and developing novel therapeutic strategies for neurological disorders.
Calcium ions (Ca²⁺) act as a ubiquitous intracellular messenger, regulating diverse cellular functions including secretion, contraction, cellular excitability, and gene expression across all organ systems [18]. The pioneering recognition that calcium ions are essential for regulating biological processes dates back to Sydney Ringer, who serendipitously found that 'lime salt' is necessary to maintain contractions of an isolated frog heart [18]. Understanding Ca²⁺ signals and their temporal and spatial characteristics in cells and tissues is crucial for elucidating physiological regulation of organ systems and for developing pharmacological approaches [18]. The development of technologies to monitor cellular Ca²⁺ signals has revolutionized our understanding of cellular signaling, enabling researchers to decode how cells use Ca²⁺ signatures to encode information specific to a given stimulus [19]. This whitepaper traces the historical progression of calcium detection methods from early biological probes to modern genetically encoded indicators, providing technical guidance for researchers investigating calcium signaling in live cells and drug discovery applications.
The Aequorin Era: Harnessing Bioluminescence from Jellyfish
The earliest measurements of intracellular Ca²⁺ dynamics began with the extraction and characterization of the Ca²⁺-sensitive bioluminescent protein aequorin from the jellyfish Aequoria victoria [18]. Aequorin is a Ca²⁺-binding photoprotein composed of an apoprotein (apoaequorin) with an approximate molecular weight of 22 kDa and a prosthetic group, a luciferin molecule called coelenterazine [19]. The protein contains three EF-hand Ca²⁺-binding sites, and when these sites are occupied by Ca²⁺, aequorin undergoes a conformational change and behaves as an oxygenase that converts coelenterazine into excited coelenteramide, which is set free together with carbon dioxide [19]. As the excited coelenteramide relaxes to the ground state, blue light (λ = 469 nm) is emitted, which can be detected with a luminometer [19].
Initial methodologies involved microinjecting aequorin extracted from jellyfish tissue into individual cells to monitor rapid changes in intracellular Ca²⁺ by measuring luminescence changes [18]. This approach was technically challenging and not widely accessible, limiting its adoption primarily to specialized physiology laboratories. The method was further constrained because aequorin is consumed during light emission, making it unsuitable for long-term measurements [18]. The development of recombinant aequorin technology represented a significant advancement, allowing researchers to address a calcium reporter-protein to specific sub-cellular compartments at will [19]. This innovation enabled measurement and comparison of calcium fluctuations occurring simultaneously in different organelles or compartments, permitting evaluation of their relative contribution to responses elicited by stimuli.
Experimental Protocol: Aequorin-Based Calcium Measurements in Plant Cells
The aequorin methodology was particularly valuable for studying calcium signatures in plant cells, where Ca²⁺ serves as a key second messenger in signaling pathways [19]. The following protocol outlines the standard procedure for aequorin-based measurements:
Cell Culture Preparation: Maintain transgenic cell suspension cultures of soybean (Glycine max L., line 6.6.12) expressing apoaequorin at 22°C under constant light conditions (3,000 lux) on a rotary shaker (125 rpm) in Murashige & Skoog medium supplemented with 5 g l⁻¹ sucrose, 1 mg l⁻¹ α-naphthylacetic acid, and 0.2 mg l⁻¹ kinetin, pH 5.8 [19]. Transformed tobacco (Nicotiana tabacum L. cv BY-2) suspension cells are grown under agitation (130 rpm) at 25°C in darkness in Linsmaier & Skoog (LS) medium supplemented with 30 g l⁻¹ of sucrose and 1 mg ml⁻¹ of 2,4-dichlorophenoxyacetic acid, pH 5.8 [19].
Nuclear Targeting: For compartment-specific measurements, construct nucleus-targeted apoaequorin using chimeric constructs including the CaMV 35S promoter that controls the nucleoplasmin coding region placed in frame with the coding region of apoaequorin [19]. Insert the whole chimeric gene into the EcoR1 site of the Agrobacterium tumefaciens binary vector pBIN19 and mobilize the plasmid from Escherichia coli to A. tumefaciens LBA4404 strain [19].
In Vivo Reconstitution: Collect cells during the exponential growth phase by filtration, wash with fresh medium, and resuspend at a 20% packed cell volume in fresh medium [19]. Perform in vivo reconstitution of aequorin by incubating an appropriate volume of washed cells with 2.5 μM coelenterazine for 4-6 hours [19].
Luminescence Measurements: Transfer coelenterazine-charged cells to a luminometer cuvette and record the basal level of luminescence for 1-2 minutes before applying stimuli [20]. For automated high-throughput applications, adapt the system to 96-well plate formats using digital luminometers capable of detecting the 469 nm emission [20].
Data Analysis: Convert luminescence data to [Ca²⁺] values using the following equation: [Ca²⁺] = ((L - Lmin)/(Lmax - L))^(1/2.5) × KD, where L is the measured light emission, Lmax is the maximum light obtained by discharging all aequorin with digitonin, Lmin is the background luminescence, and KD is the apparent dissociation constant [19].
Figure 1: Aequorin calcium detection mechanism workflow
Tsien's Synthetic Dyes: Revolutionizing Calcium Imaging
The field of calcium signaling experienced a transformative advancement with Roger Tsien's pioneering development of synthetic fluorescent Ca²⁺ indicators in the 1970s and 1980s [18]. These indicators were based on the Ca²⁺ chelating properties of EGTA and BAPTA (1,2-bis(o-aminophenoxy)ethane-N,N,N',N'-tetraacetic acid), which provided selective binding sites for calcium ions [21]. The most significant innovation was the coupling of these chelators to fluorophores, creating molecules whose spectral properties changed upon Ca²⁺ binding [21]. This breakthrough enabled direct monitoring of cellular Ca²⁺ signals with high temporal and spatial resolution, making calcium imaging accessible to a broad scientific community.
The earliest synthetic indicators included Quin-2, but the field rapidly expanded to include Fura-2, Indo-1, Fluo-3, and Fluo-4, each with distinct spectral properties and calcium affinities suitable for different experimental applications [22]. These indicators could be loaded into cells using membrane-permeant AM ester derivatives, which are hydrolyzed by intracellular esterases to release the active, membrane-impermeant indicator [21]. This loading strategy eliminated the need for microinjection and enabled population-level studies of calcium dynamics, significantly expanding the experimental possibilities for investigating calcium signaling in diverse cell types.
Technical Classification of Synthetic Calcium Indicators
Synthetic calcium indicators can be categorized based on their spectral response mechanisms and properties:
Ratiometric Indicators: These indicators undergo a shift in either excitation or emission wavelength upon calcium binding. Fura-2 exhibits excitation wavelength shifts (363/335 nm to 363/335 nm with emission at 512/505 nm), while Indo-1 shows emission wavelength shifts (482/398 nm with excitation at 349/331 nm) [22] [21]. Ratiometric measurements provide internal controls that minimize artifacts from uneven dye loading, photobleaching, or variations in cell thickness [22] [21].
Non-Ratiometric (Single-Wavelength) Indicators: This class includes Fluo-3, Fluo-4, and Oregon Green derivatives, which exhibit increased fluorescence intensity upon calcium binding without spectral shifts [22]. Fluo-4, for example, displays a >100-fold increase in fluorescence intensity with excitation/emission at 494/506 nm [22]. While these indicators require careful calibration for quantitative measurements, they offer high sensitivity and are compatible with standard FITC filter sets [22].
Table 1: Properties of Major Synthetic Calcium Indicators
| Indicator | Ratiometric | Excitation (nm) | Emission (nm) | Kd (nM) | Signal Change | Primary Applications |
|---|---|---|---|---|---|---|
| Fura-2 | Yes (Ex) | 363/335 | 512/505 | 145 | Shift in excitation ratio | Quantitative [Ca²⁺] measurement |
| Indo-1 | Yes (Em) | 349/331 | 482/398 | 230 | Shift in emission ratio | Flow cytometry, quantitative imaging |
| Fluo-3 | No | 506 | 525 | 390 | >100-fold intensity increase | High-sensitivity detection |
| Fluo-4 | No | 494 | 506 | 335 | >100-fold intensity increase | HTS, confocal microscopy |
| Oregon Green 488 BAPTA-1 | No | 494 | 523 | 170 | 14-fold intensity increase | Standard fluorescence detection |
| Rhod-2 | No | 556 | 576 | 1000 | 50-fold intensity increase | Mitochondrial localization |
Experimental Protocol: Loading and Imaging with Synthetic Indicators
The standard methodology for using synthetic calcium indicators involves several critical steps:
Indicator Selection: Choose an indicator based on experimental needs. For quantitative measurements, select ratiometric indicators (Fura-2, Indo-1). For high-sensitivity detection of small changes, choose high-dynamic-range indicators (Fluo-4, Fluo-3) [22] [21]. Consider the equipment available—Fura-2 requires dual-excitation capability, while Fluo-4 works with standard confocal microscopes [22].
AM Ester Preparation: Prepare stock solutions of cell-permeant AM ester forms in anhydrous DMSO at 1-5 mM concentration [21]. Use Pluronic F-127 (0.01-0.1%) to improve dye solubility and loading efficiency, particularly for hydrophobic indicators [21].
Cell Loading: Incubate cells with 1-10 μM indicator AM ester in physiological buffer for 30-60 minutes at 20-37°C [22] [21]. Include probenecid (1-2.5 mM) for cell types with active organic anion transport that might export the dye [22].
De-esterification: Allow loaded cells to remain in dye-free buffer for 15-30 minutes to ensure complete hydrolysis of AM esters by intracellular esterases [21].
Calibration (For Quantitative Measurements): For ratiometric indicators, perform in situ calibration using ionophores (ionomycin or A-23187) in calcium-free (with EGTA) and high-calcium buffers to determine Rmin and Rmax [21]. Calculate [Ca²⁺] using the Grynkiewicz equation: [Ca²⁺] = Kd × (R - Rmin)/(Rmax - R) × (Sf2/Sb2) [18].
Figure 2: Synthetic dye loading and calcium detection pathway
Genetically Encoded Calcium Indicators (GECIs): The Molecular Biology Revolution
The advent of genetically encoded calcium indicators (GECIs) represented a paradigm shift in calcium imaging, addressing key limitations of synthetic dyes while introducing new capabilities for cell-specific targeting and long-term monitoring. GECIs are engineered fusion proteins that typically combine a calcium-binding domain (such as calmodulin or troponin C) with fluorescent protein pairs (for FRET-based sensors) or single circularly permuted fluorescent proteins (for intensiometric sensors) [23]. The earliest GECIs included cameleons (FRET-based) and camgaroos/G-CaMPs (single-FP based), which have evolved through successive generations into the high-performance indicators available today [23].
The GCaMP series has emerged as particularly impactful, with GCaMP6f and related variants enabling detection of single action potentials in neurons [24]. These indicators are based on circularly permuted green fluorescent protein (cpGFP), calmodulin (CaM), and the M13 peptide fragment of myosin light chain kinase [24]. Calcium binding to CaM promotes interaction with M13, leading to conformational changes that enhance the fluorescence of cpGFP [24]. More recent developments have focused on red and far-red GECIs, such as the FR-GECO series, which offer advantages including reduced phototoxicity, lower autofluorescence, better tissue penetration, and spectral compatibility with optogenetic tools [23].
Advanced GECIs: Far-Red Indicators and Spectral Diversity
The latest generation of GECIs has expanded into longer wavelengths, with recently developed FR-GECO1a and FR-GECO1c exhibiting excitation/emission maxima at approximately 596/642 nm and 596/646 nm, respectively [23]. These far-red indicators are based on the monomeric far-red fluorescent proteins mKelly1 and mKelly2, offering large responses to Ca²⁺ (ΔF/F0 = 6 for FR-GECO1a, 18 for FR-GECO1c) and high affinities (apparent Kd = 29 nM for FR-GECO1a, 83 nM for FR-GECO1c) [23]. Their spectral properties within the optical window (600-1300 nm) enable deeper tissue imaging and multicolor experiments with other optogenetic indicators and actuators [23].
Table 2: Comparison of Modern Genetically Encoded Calcium Indicators
| GECI | Type | Excitation (nm) | Emission (nm) | Kd (nM) | ΔF/F0 (%) | Response Kinetics | Primary Applications |
|---|---|---|---|---|---|---|---|
| GCaMP6f | Green cpGFP | 488 | 510 | 145 | ~700 | Fast | Neuronal imaging, in vivo monitoring |
| jRGECO1a | Red RFP-based | 558 | 580 | 160 | ~500 | Medium | Multiplexing with optogenetics |
| FR-GECO1a | Far-red | 596 | 642 | 29 | 600 | Medium | Deep tissue imaging, multiplexing |
| FR-GECO1c | Far-red | 596 | 646 | 83 | 1800 | Medium | High-contrast deep tissue imaging |
| NIR-GECO1 | Near-infrared | 678 | 704 | 215 | -1000 (inverted) | Slow | Maximum tissue penetration |
Experimental Protocol: Implementation of GECIs for In Vivo Imaging
The application of GECIs requires molecular biology techniques for expression in target cells or organisms:
Vector Construction: Clone GECI coding sequences into appropriate expression vectors under cell-specific promoters for targeted expression [24]. For stable expression, use systems such as the AAVS1 safe harbor locus in human iPSCs with TALEN-based integration [24].
Cell Transfection/Transduction: Deliver genetic constructs to target cells using appropriate methods (lipofection, electroporation, viral transduction). For difficult-to-transfect cells, use lentiviral or adenoviral vectors for efficient gene delivery [23].
Stable Cell Line Generation: For long-term studies, generate stable cell lines through antibiotic selection or fluorescence-activated cell sorting (FACS) of high-expressing cells [24]. The MHHi001-A-5 iPSC line exemplifies this approach, with GCaMP6f and RedStar nuc reporters integrated into the AAVS1 locus [24].
In Vivo Imaging: For live animal imaging, use two-photon microscopy for deep tissue penetration with minimal scattering [18]. Implement fiber-optic-based microendoscopes for imaging in awake, behaving animals [18]. Maintain specimens under appropriate physiological conditions during imaging.
Data Analysis: Process time-series image data to extract fluorescence traces (ΔF/F0). For neuronal applications, use spike inference algorithms to detect action potentials from calcium transients [23].
Figure 3: GECI implementation and experimental workflow
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful calcium imaging requires appropriate selection of reagents and materials tailored to specific experimental needs. The following table summarizes key components for calcium signaling research:
Table 3: Essential Research Reagents for Calcium Signaling Studies
| Reagent Category | Specific Examples | Function/Application | Key Considerations |
|---|---|---|---|
| Calcium Indicators | Aequorin, Fura-2 AM, Fluo-4 AM, Rhod-2 AM, GCaMP6f | Direct detection of calcium ions | Select based on sensitivity, targeting, and equipment compatibility |
| Calmodulin Ligands | W-7, calmidazolium, trifluoperazine | Inhibition of calmodulin-dependent processes | Useful for dissecting calcium signaling pathways |
| Calcium Chelators | BAPTA AM, EGTA, 5,5'-dimethyl BAPTA | Control of intracellular calcium concentration | BAPTA has faster kinetics than EGTA |
| Ionophores | Ionomycin, A-23187 (calcimycin) | Equalize calcium across membranes for calibration | Use for maximum and minimum signal determination |
| Calcium Buffer Systems | Calcium Calibration Buffer Kit | Precise calcium concentrations for calibration | Essential for quantitative measurements |
| Pluronic F-127 | Non-ionic surfactant | Improves AM ester solubility and loading | Critical for hydrophobic indicators |
| Esterase Inhibitors | Probenecid | Reduces dye export in certain cell types | Extends dye retention time |
| Caged Calcium Compounds | DMNP-EDTA (DM-Nitrophen) | UV-triggered calcium release | Enables precise temporal control of calcium pulses |
The evolution from aequorin to Tsien's dyes and onward to modern GECIs represents a remarkable technological journey that has fundamentally transformed our understanding of calcium signaling biology. Each advancement has addressed limitations of previous approaches while introducing new capabilities: aequorin enabled the first intracellular calcium measurements, synthetic dyes provided accessibility and ratiometric quantification, and GECIs offered genetic targeting and long-term monitoring. Current research focuses on expanding the spectral range of indicators, improving signal-to-noise ratios, developing innovative targeting strategies, and creating multifunctional sensors that simultaneously monitor calcium alongside other physiological parameters. The integration of these advanced calcium detection technologies with cutting-edge imaging platforms and optogenetic tools continues to drive discoveries in basic physiology and drug development, promising new insights into the complex spatiotemporal language of cellular calcium signaling.
Calcium (Ca²⁺) is the most ubiquitous signaling molecule in biology, acting as a critical second messenger that regulates processes ranging from neurotransmitter release in milliseconds to developmental processes over days [25]. The ability to detect and quantify these spatial and temporal fluctuations in calcium concentration is fundamental to modern cell biology and neuroscience. Calcium imaging techniques, which optically measure the calcium status of cells and tissues, rely on fluorescent molecules that change their optical properties upon binding Ca²⁺ ions [26]. These calcium indicators transduce the chemical event of calcium binding into a measurable fluorescent signal, allowing researchers to visualize signaling dynamics in real-time within living cells. This technical guide examines the core operating principles of these molecular tools, which are indispensable for deciphering the complex language of calcium signaling in physiological and pathological contexts.
Fundamental Principles of Fluorescent Calcium Indicators
Core Mechanism of Fluorescence Transduction
All fluorescent calcium indicators operate on the same fundamental principle: they contain a calcium-binding moiety that undergoes a conformational change upon Ca²⁺ coordination, which in turn alters the electronic environment of a coupled fluorophore. This alteration manifests as a change in the fluorophore's fluorescence properties. For chemical indicators, the calcium-binding moiety is typically a synthetic chelator based on BAPTA (a derivative of EGTA), which provides high selectivity for Ca²⁺ over other biologically relevant ions like magnesium (Mg²⁺) [26]. Genetically encoded indicators, in contrast, often utilize natural calcium-binding proteins such as calmodulin (CaM) or troponin C as their sensing domain [25] [26]. The key performance parameters for any calcium indicator are its binding affinity (Kd), which determines the dynamic range of calcium concentrations it can report, and its kinetics (kon and koff rates), which determine its ability to track rapid calcium transients [27].
The Critical Role of Indicator Affinity and Kinetics
The binding affinity of an indicator, expressed as the dissociation constant (Kd), must be carefully matched to the expected calcium concentration range in the cellular compartment being studied. High-affinity indicators (Kd in the nanomolar range, e.g., Fura-2, GCaMP) are ideal for measuring resting cytosolic calcium or small fluctuations, whereas low-affinity indicators (Kd in the micromolar to millimolar range, e.g., Fluo-5N, NEMOer variants) are essential for monitoring calcium in compartments with high basal calcium levels, such as the endoplasmic reticulum (ER) or sarcoplasmic reticulum (SR) [25] [27] [28]. The kinetics of the indicator, particularly the calcium dissociation rate (koff), dictates the temporal resolution. For example, to capture elementary calcium release events like calcium sparks and calcium blinks in muscle cells, indicators require koff rates exceeding 1000 s⁻¹ [25]. Recent advances in sensor design, such as the NEMOer-f indicator with a koff of 156.75 s⁻¹, have enabled the inaugural detection of SR calcium blinks in cardiomyocytes, events that were previously too fast to capture [28].
Chemical Calcium Indicators
Operating Principles and Classes
Chemical indicators are small, synthetic molecules that function as Ca²⁺ chelators linked to a fluorophore. The binding of Ca²⁺ to the chelator directly affects the photophysical properties of the fluorophore. These indicators are typically loaded into cells in an acetoxymethyl (AM) ester form, which is lipophilic and cell-permeant. Once inside the cell, endogenous esterases cleave the AM ester groups, trapping the active, charged indicator in the cytosol [26]. Chemical indicators are broadly categorized into two classes based on their spectral behavior upon calcium binding.
Table 1: Common Chemical Calcium Indicators and Their Properties
| Indicator | Type | Excitation (nm) | Emission (nm) | Kd (nM) | Primary Applications |
|---|---|---|---|---|---|
| Fluo-4 | Single wavelength | ~490 | ~520 | 345 [27] | Fast confocal imaging, high-throughput screening |
| X-rhod-1 | Single wavelength | ~580 | ~600 | 700 [27] | Multiplexing with other probes, reduced autofluorescence |
| Fura-2 | Ratiometric | 340/380 | ~510 | 145 [27] | Precise quantitative calcium measurement in cytosol |
| Indo-1 | Ratiometric | ~340 | 405/485 | 230 [27] | Flow cytometry, quantitative kinetic studies |
| Calcium Green-5N | Single wavelength | ~506 | ~533 | ~14,000 [25] | Measuring high [Ca²⁺] in organelles (e.g., ER, SR) |
Single Wavelength Indicators
Fluo-4 is a quintessential single wavelength indicator. Its core operating principle is fluorescence intensiometry—upon binding Ca²⁺, it exhibits a significant increase in fluorescence intensity (often >100-fold) without a substantial shift in its excitation or emission spectra [27]. The mechanism involves the Ca²⁺-bound form of the chelator suppressing a photo-induced electron transfer (PeT) quenching pathway that is active in the Ca²⁺-free state, leading to dramatic brightening. The major advantage of Fluo-4 is its convenience and compatibility with standard fluorescein filter sets and 488 nm laser lines on confocal microscopes. However, its intensiometric nature makes quantification susceptible to artifacts from variations in dye concentration, photobleaching, and cell thickness, as the absolute signal depends on the baseline fluorescence, F(0) [27].
Ratiometric Indicators
Fura-2 and Indo-1 are ratiometric indicators that provide a more robust quantitative measurement. Their operating principle involves a Ca²⁺-dependent spectral shift.
- Fura-2 exhibits a shift in its excitation spectrum. When Ca²⁺-free, it is best excited at ~380 nm, but upon Ca²⁺ binding, the excitation maximum shifts to ~340 nm, with the emission remaining at ~510 nm [27]. The ratio of fluorescence (510 nm emission) when excited at 340 nm versus 380 nm is directly related to the Ca²⁺ concentration. This ratio is self-correcting for many artifacts, including dye concentration and path length.
- Indo-1 operates via a shift in its emission spectrum. With excitation at ~340 nm, the emission maximum shifts from ~485 nm (Ca²⁺-free) to ~405 nm (Ca²⁺-bound) [27]. The ratio of emission at 405 nm to 485 nm is used for calculation. While powerful, ratiometric imaging requires more complex instrumentation, including multiple excitation or emission wavelengths and rapid switching systems [27].
Figure 1: Fura-2 Ratiometric Transduction Principle. The indicator's preferred excitation wavelength shifts from 380 nm to 340 nm upon calcium binding, enabling quantitative ratio measurement.
Genetically Encoded Calcium Indicators (GECIs)
Fundamental Design and Transduction Strategies
GECIs are engineered fluorescent proteins whose fluorescence is modulated by inter- or intra-molecular interactions that are sensitive to Ca²⁺. They are encoded by genes that can be transfected into cells or expressed in transgenic organisms, allowing for long-term studies, targeting to specific cell types or subcellular compartments, and in vivo imaging [26]. The Ca²⁺ sensing is predominantly achieved through the integration of calmodulin (CaM) and its target peptide M13, or the calcium-binding protein troponin C (TnC). The fluorescence reporting occurs via two primary mechanistic classes: single-FP sensors and FRET-based paired-FP sensors.
Table 2: Major Classes of Genetically Encoded Calcium Indicators (GECIs)
| GECI Class/Name | Sensing Mechanism | Reporting Mechanism | Key Characteristics | Example Kd / Kinetics |
|---|---|---|---|---|
| GCaMP (e.g., GCaMP6) | CaM-M13 | cpGFP (intensiometric) | High affinity, bright, widely used in neuroscience [26] | Kd ~235 nM [27] |
| Cameleon | CaM-M13 | FRET pair (e.g., CFP/YFP) | Ratiometric, good for quantification [26] | Varies with construct |
| TN-XXL | Troponin C | FRET pair (Cerulean/Citrine) | Reduced interference with endogenous CaM signaling [25] | koff ~1.1 s⁻¹ [25] |
| NEMOer-f | Engineered CaM | mNeonGreen (intensiometric) | Low affinity for ER/SR, very fast kinetics [28] | Kd ~mM range, koff = 156.75 s⁻¹ [28] |
| RCaMP/jRGECO1 | CaM-M13 | Red fluorescent protein (mRuby/mApple) | Multiplexing, deeper tissue penetration [26] | Varies with construct |
Single-Fluorescent Protein Sensors (e.g., GCaMP, NEMOer)
The GCaMP family represents the most widely used single-FP GECIs. Its core design is a circularly permuted green fluorescent protein (cpGFP) sandwiched between calmodulin (CaM) and a M13 peptide (a CaM-binding domain) [26]. In the low Ca²⁺ state, the cpGFP chromophore is protonated and less fluorescent. Binding of Ca²⁺ to CaM promotes its interaction with M13, causing a conformational change that stabilizes the deprotonated state of the cpGFP chromophore, resulting in a large increase in green fluorescence. The NEMOer sensors represent a recent advancement for imaging high calcium environments like the ER/SR. They are engineered from mNeonGreen and incorporate mutations in the CaM domain that significantly lower Ca²⁺ affinity (to the millimolar range) and dramatically increase the dynamic range (up to 80-fold greater than previous sensors like G-CEPIA1er) [28]. This is achieved by increasing the proportion of the fluorophore that exists in a bright, anionic state when Ca²⁺ is bound.
Figure 2: Single-FP GECI (GCaMP) Transduction. Calcium binding induces a conformational change that deprotonates the chromophore, increasing fluorescence.
Förster Resonance Energy Transfer (FRET) Sensors (e.g., Cameleon)
Cameleon sensors are the prototypical FRET-based GECIs. They consist of a CFP donor and a YFP acceptor fluorophore flanking a central sensing module composed of CaM and the M13 peptide [26]. In the absence of Ca²⁺, the sensing module is extended, separating the two FPs and minimizing FRET efficiency; excitation of CFP results primarily in CFP emission. When Ca²⁺ binds to CaM, it wraps around M13, bringing the CFP and YFP into close proximity. This spatial change allows for efficient FRET: upon CFP excitation, energy is non-radiatively transferred to YFP, resulting in predominant YFP emission. The ratio of YFP emission to CFP emission provides a quantitative, ratiometric measure of Ca²⁺ concentration that is largely insensitive to sensor concentration and excitation light intensity.
Experimental Considerations and Protocols
Selecting the Appropriate Indicator
Choosing the correct indicator is critical for experimental success and depends on several factors:
- Cellular Compartment: For cytosolic measurements (
100 nM resting), high-affinity sensors like Fura-2 or GCaMP are suitable. For ER/SR measurements (100-500 μM), low-affinity sensors like NEMOer, Mag-Fura-2, or Fluo-5N are essential [25] [27] [28]. - Kinetic Requirements: For fast neuronal spiking or muscle contraction events, indicators with fast on/off rates (e.g., R-CatchER, koff >2100 s⁻¹) are needed to avoid distorting transient kinetics [25].
- Quantification Needs: Ratiometric indicators (Fura-2, Cameleon) are superior for precise quantification, while intensiometric indicators (Fluo-4, GCaMP) are simpler and often brighter.
- Multiplexing & Instrumentation: Red-shifted indicators (X-rhod, RCaMP) help reduce autofluorescence and allow multiplexing with other probes. Instrument capability (e.g., UV lasers for Fura-2) can be a limiting factor [27].
Key Experimental Protocol: Live-Cell Calcium Imaging in a Flow System
The following protocol, adapted from a 2025 study, outlines a robust procedure for measuring calcium responses to shear stress in endothelial cells, demonstrating key steps applicable to many live-cell imaging experiments [29].
- Cell Culture and Preparation: Culture human lung microvascular endothelial cells (HMVEC-Ls) in complete EGM-2 medium until 70-90% confluent. Use cells between passages 3-7 for experimental consistency.
- Microfluidic Device Seeding: Seed the cells into an Ibidi Luer VI microfluidic channel at an appropriate density and allow them to adhere and form a monolayer under static conditions for 24-48 hours.
- Dye Loading: On the day of imaging, prepare a working solution of the cell-permeant calcium indicator (e.g., Fluo-8 AM). Replace the culture medium in the reservoir with the dye solution. Incubate the cells in the dark at room temperature for 30-60 minutes to allow dye loading and esterase cleavage.
- System Setup and Perfusion: Connect the microfluidic device to a peristaltic pump via sterilized tubing. Wash out excess dye by perfusing with pre-warmed Hanks' Balanced Salt Solution (HBSS) or imaging buffer for at least 15 minutes to establish a stable baseline.
- Image Acquisition: Place the device on a microscope stage with a maintained environment (e.g., 37°C, 5% CO₂). Use a 20x objective. For Fluo-8 (similar to Fluo-4), use 488 nm excitation and collect emission at ~520 nm. Acquire time-series images at a frame rate sufficient to capture the expected calcium dynamics (e.g., 1-10 frames per second).
- Stimulation and Data Collection: Initiate the application of defined shear stress via the pump while continuously acquiring images. Include control conditions without stimulation.
- Data Analysis: Analyze the fluorescence changes over time (F(t)) for individual cells or regions of interest (ROI). Normalize the data to the baseline fluorescence (F₀) and express as ΔF/F₀. Use analysis software (e.g., ImageJ plugins, CalciumNetExploreR) for trace extraction, event detection, and statistical analysis [29] [14].
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for Calcium Imaging
| Item | Function/Description | Example Use Case |
|---|---|---|
| Fluo-8 AM / Fluo-4 AM | Cell-permeant, single-wavelength chemical indicator. Fluo-8 offers brighter signal and room temperature loading [29]. | General cytosolic calcium imaging in response to pharmacological or physical stimuli. |
| Fura-2 AM | Cell-permeant, ratiometric chemical indicator for quantitative measurement. | Precise quantification of cytosolic [Ca²⁺] in response to slow-onset agonists. |
| GCaMP6/GCaMP8 AAV | Genetically encoded calcium indicator delivered via Adeno-Associated Virus. | Long-term calcium imaging in specific neuronal populations in vivo or in primary culture. |
| NEMOer Plasmid DNA | Genetically encoded low-affinity indicator targeted to the ER/SR [28]. | Imaging calcium storage and release dynamics in the endoplasmic or sarcoplasmic reticulum. |
| Ionomycin | Calcium ionophore used to maximally increase intracellular calcium. | Calibration of fluorescence signal or positive control to ensure indicator functionality. |
| Pluronic F-127 | Non-ionic surfactant that disperses AM esters in aqueous solution. | Aiding the loading of hydrophobic AM-ester dyes into cells. |
| CoolLED pE-340fura | LED light source with dedicated 340 nm and 380 nm outputs. | Enabling stable, high-speed ratiometric imaging with Fura-2 without the use of arc lamps [27]. |
| CalciumNetExploreR (R Package) | Software for downstream analysis of extracted calcium traces [14]. | Network analysis, functional connectivity mapping, and population activity analysis from time-series data. |
The transduction of calcium binding into a fluorescent signal is achieved through elegantly engineered molecular mechanisms, from the simple chelation-induced brightening of Fluo-4 to the complex CaM-mediated conformational changes in GCaMP and the proximity-dependent FRET in Cameleon. The continuous evolution of these tools—toward faster kinetics, broader dynamic ranges, more colors, and better targeting—is pivotal for advancing our understanding of calcium signaling in health and disease. The choice of indicator and experimental protocol must be carefully aligned with the biological question, whether it involves mapping neuronal networks at millisecond resolution or quantifying subtle perturbations in organellar homeostasis over minutes. As the field progresses, the integration of these sophisticated molecular reporters with advanced imaging modalities and analysis software will undoubtedly unveil new dimensions of calcium signaling complexity.
Calcium ions (Ca²⁺) are universal intracellular messengers that regulate a plethora of cellular processes, including signal transduction, neurotransmitter release, muscle contraction, enzyme cofactor activity, and fertilization [30]. Analyzing calcium flux using live cell imaging techniques is therefore fundamental to understanding both normal cellular function and dysfunctions underlying disease states [30]. The development of diverse calcium indicators—each with unique properties and applications—has created a rich ecosystem of tools for researchers. This ecosystem primarily comprises chemical dyes, genetically encoded calcium indicators (GECIs), and luminescent reporters, all enabling the visualization of calcium dynamics in living cells with high spatiotemporal resolution. The choice of indicator is critical and depends on the specific biological question, experimental model, and methodological constraints.
Chemical Calcium Indicators
Chemical calcium indicators are small, synthetic molecules that bind calcium ions via chelation. Most are based on calcium chelators like BAPTA (1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid), which offers enhanced calcium specificity and pH stability compared to EGTA [30]. To facilitate cell loading, these indicators are often modified with acetoxymethyl (AM) esters, rendering them lipophilic and cell-permeant. Once inside the cell, endogenous esterases cleave the AM esters, freeing the carboxyl groups and trapping the active indicator inside, where it can bind Ca²⁺ [30] [27]. Binding typically results in either an increase in the quantum yield of fluorescence or a shift in emission or excitation wavelength.
Table 1: Properties of Common Chemical Calcium Indicators
| Indicator | Type | Excitation (nm) | Emission (nm) | Kd (nM) | Key Features | Primary Applications |
|---|---|---|---|---|---|---|
| Fura-2 [27] | Ratiometric | 340/380 | ~510 | 145 | Absorption shift with Ca²⁺; robust to artifacts | Quantitative microscopy (ratio-imaging) |
| Indo-1 [30] [27] | Ratiometric | ~340 | 400/475 | 230 | Emission shift with Ca²⁺; preferred for flow cytometry | Flow cytometry, quantitative microscopy |
| Quin-2 [30] | Intensity-based | 339 | 492 | High affinity | High selectivity; sensitive for low calcium levels | Measuring resting calcium levels |
| Fluo-4 [30] [27] [22] | Intensity-based | 490 | 520 | 345 | >100-fold fluorescence increase; no resting signal | Confocal microscopy, flow cytometry, HTS |
| Oregon Green BAPTA-1 [22] | Intensity-based | 494 | 523 | ~170 | 14-fold increase; visible signal at rest | Cell location visualization prior to stimulation |
| Rhod-2 [22] | Intensity-based | 552 | 581 | ~570 | 50-fold increase; localizes to mitochondria | Multiplexing with GFP; mitochondrial calcium |
| X-rhod-1 [27] | Intensity-based | 580 | 600 | 700 | Long-wavelength excitation and emission | Reduced phototoxicity, multiplexing |
Experimental Protocol: Measuring Calcium Transients with Fluo-4 AM
This protocol is widely used for detecting agonist-stimulated calcium flux in live cells, suitable for high-throughput screening and confocal microscopy [22].
- Dye Preparation: Prepare a 1-5 µM working solution of Fluo-4 AM in a physiological buffer (e.g., Hanks' Balanced Salt Solution, HBSS) or serum-free culture medium. The use of phenol red-free media is recommended to reduce background fluorescence [31].
- Cell Loading: Replace the culture medium on the adherent cells with the dye working solution. Incubate for 30-60 minutes at 37°C or room temperature, protected from light.
- Dye Desterification and Washing: After incubation, remove the dye solution and wash the cells with fresh buffer to remove extracellular dye. Subsequently, incubate the cells for an additional 20-30 minutes in dye-free buffer to allow complete cleavage of the AM esters by intracellular esterases, activating the indicator.
- Image Acquisition: Place the cells on the microscope stage, maintaining environmental control (37°C, 5% CO₂ if possible). For Fluo-4, use standard FITC/GFP filter sets (excitation ~490 nm, emission ~520 nm). Acquire baseline images for 30-60 seconds to establish the resting fluorescence (F₀).
- Stimulus Application: Introduce the agonist or stimulus of interest (e.g., histamine, ATP) without interrupting the imaging sequence. The addition of 1 µM histamine, for instance, can induce calcium oscillations, while 5 µM ionomycin can be used to induce maximal calcium influx [30].
- Data Analysis: Quantify the fluorescence changes over time. Data is often expressed as ΔF/F₀, where ΔF is the change in fluorescence from baseline, and F₀ is the baseline fluorescence [27].
Genetically Encoded Calcium Indicators (GECIs)
GECIs are engineered proteins that typically fuse a fluorescent protein (FP) to a calcium-sensing domain, such as calmodulin (CaM) and its target peptide. Their major advantage is the ability to be genetically targeted to specific cell types, subcellular compartments, or within transgenic animal models, enabling long-term and in vivo studies [32] [33] [34]. While early GECIs had limitations in brightness and kinetics, recent developments have yielded indicators with improved performance.
Table 2: Properties of Representative Genetically Encoded Calcium Indicators
| Indicator | Class / Color | Excitation (nm) | Emission (nm) | Apparent Kd | ΔF/F0 (%) | Key Features & Applications |
|---|---|---|---|---|---|---|
| GCaMP6s [35] | Green, single FP | ~480 | ~510 | ~235 nM | High | High affinity; detection of single action potentials in neurons |
| FR-GECO1a [33] | Far-Red, single FP | ~596 | ~642 | 29 nM | 500% (6-fold) | Far-red emission within optical window; multiplexing |
| FR-GECO1c [33] | Far-Red, single FP | ~596 | ~646 | 83 nM | 1700% (18-fold) | High contrast; bright under one- and two-photon illumination |
| MaPCa Dyes [32] | Rhodamine-based, tunable | 558-656 | 580-670 | 410 nM - 457 µM | 500-1100% | Targetable via HaloTag; no-wash imaging; tuneable affinity |
| Cameleon [27] | Ratiometric, FRET-based | Donor-dependent | Donor/Acceptor | Varies | Ratiometric FRET change | Ratiometric readout; reduced susceptibility to artifacts |
Experimental Protocol: Lentiviral Transduction for Stable GECI Expression
This protocol describes how to establish a cell line stably expressing a GECI, such as GCaMP6s, for long-term calcium imaging studies [31].
- Virus Production and Transduction: Produce a lentivirus encoding the GECI (e.g., GCaMP6s) following standard biosafety protocols. Apply the lentivirus to the target cells (e.g., MA104 monkey kidney cells, neuronal cultures, or organoids) at an appropriate multiplicity of infection (MOI) in the presence of a transduction-enhancing agent like Polybrene.
- Selection and Expansion: After 24-48 hours, replace the virus-containing medium with fresh growth medium, potentially supplemented with an antibiotic (e.g., puromycin) if the vector contains a selectable marker, to select for successfully transduced cells. Expand the positive population.
- Validation: Validate GECI expression using fluorescence microscopy or flow cytometry before proceeding to experiments.
- Live-Cell Imaging: Plate the transduced cells on an appropriate imaging vessel, such as a glass-bottom dish or µ-Slide. For imaging, use phenol red-free media and maintain environmental control. For GCaMP6s, image using standard GFP filter sets (excitation ~470/22 nm, emission ~510/42 nm) [31] [36].
- Data Acquisition and Analysis: Capture images at regular intervals (e.g., every 1-2 seconds for fast neuronal activity or every minute for long-term processes like infection). Analyze the time series data to detect calcium transients, often defined as fluorescence increases exceeding a set threshold above the baseline noise [35].
Luminescent and Label-Free Reporters
While fluorescent indicators dominate the field, alternative approaches offer unique advantages.
Luminescent Reporters
The protein aequorin, isolated from jellyfish, is a classic luminescent calcium reporter. It emits blue light (~469 nm) upon binding Ca²⁺ in the presence of its cofactor, coelenterazine. Aequorin is highly sensitive and generates virtually no background, making it suitable for detecting very low calcium concentrations. However, its low light output often requires highly sensitive detectors [35].
Label-Free Sensing
Emerging technologies aim to detect calcium dynamics without any labels, thereby avoiding potential toxicity, photobleaching, and perturbation of native biology. One recent advancement is a gallium nitride (GaN)-based photonic microchip. This platform detects calcium-induced changes in the cellular refractive index, allowing for label-free, long-term monitoring of single-cell calcium signaling and revealing cell-to-cell heterogeneity [37].
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Reagents and Materials for Calcium Imaging Experiments
| Reagent / Material | Function / Description | Example Use Cases |
|---|---|---|
| Fluo-4 AM Kit [36] [22] | Cell-permeant, single-wavelength chemical indicator. Kits often include probenecid to inhibit anion transport. | Short-term kinetic measurements of GPCR signaling in high-throughput formats. |
| CellLight BacMam 2.0 [36] | Baculovirus-based delivery system for labeling organelles or expressing biosensors in mammalian cells, including hard-to-transfect cells. | Efficient, stable expression of GECIs with low cellular toxicity. |
| HaloTag MaPCa Dyes [32] | Synthetic fluorogenic calcium indicators that become fluorescent upon binding to HaloTag fusion proteins. | No-wash, subcellularly targeted calcium imaging with customizable affinity. |
| CoolLED pE-340fura [27] | LED light source specifically designed for ratiometric imaging, providing stable 340 nm and 380 nm excitation. | Quantitative Ca²⁺ measurement with Fura-2, minimizing photobleaching. |
| Gibco FluoroBright DMEM [36] | Phenol red-free culture medium specially formulated to reduce background fluorescence during live-cell imaging. | All long-term live-cell imaging experiments to improve signal-to-noise ratio. |
| EVOS Onstage Incubator [36] | Microscope-stage incubator that maintains stable temperature, humidity, and CO₂ levels during time-lapse imaging. | Kinetic imaging over hours to days for processes like viral infection or wound healing. |
Visualization of Calcium Indicator Mechanisms and Workflows
Diagram 1: Calcium Indicator Workflows. This diagram contrasts the primary mechanisms for chemical dyes (top, blue) and GECIs (bottom, green) in live cells.
A Practical Guide to Calcium Imaging: From Sensor Selection to In Vivo Application
Calcium ions (Ca²⁺) are ubiquitous intracellular messengers, regulating processes from neurotransmitter release and muscle contraction to gene expression and cell death [38]. Analyzing calcium flux using live cell imaging is therefore fundamental to understanding cellular function and dysfunction in disease. The cornerstone of this field is the choice between two primary classes of molecular probes: synthetic fluorescent dyes and genetically encoded calcium indicators (GECIs). This decision is critical, as it influences experimental design, data interpretation, and the very biological questions one can address. Framed within the broader context of calcium signaling detection methods, this guide provides a structured framework to help researchers and drug development professionals select the optimal probe for their specific application, drawing on the latest advances in the field.
Core Principles and Evolution of Calcium Indicators
The Fundamental Mechanism of Calcium Detection
Both synthetic dyes and GECIs operate on the principle of fluorescent reporting. They bind to calcium ions, causing a change in their fluorescent properties—typically an increase in intensity (fluorescence intensity) or a shift in their excitation or emission wavelengths (ratiometric properties) [38] [39]. This change in fluorescence serves as a proxy for changes in intracellular Ca²⁺ concentration. A key advantage of Ca²⁺ as a signal is its low basal intracellular concentration (∼50–100 nM), which rises dramatically during an action potential, providing a high signal-to-noise ratio for detecting neuronal activity [40].
A Brief Historical Context
The development of calcium indicators has been a journey of continuous innovation. Early work in the 1960s involved injecting the photoprotein aequorin into single cells [41]. A major breakthrough came with the synthesis of the first chemical dyes, such as Quin-2, followed by Fura-2, Indo-1, and Fluo-3, which offered improved selectivity and fluorescence properties [41] [38]. The subsequent advent of GECIs, engineered by fusing fluorescent proteins with calcium-binding motifs like calmodulin, revolutionized the field by enabling genetic targeting to specific cell types and long-term longitudinal studies [40] [42]. Ongoing engineering efforts continue to yield indicators with improved brightness, dynamic range, kinetics, and spectral properties, such as the recent far-red FR-GECOs [33] and highly dynamic ER-targeted NEMOer sensors [43].
Quantitative Comparison: Synthetic Dyes vs. GECIs
The following tables summarize the key characteristics of synthetic dyes and GECIs to facilitate a direct comparison.
Table 1: Core Characteristics and Application Fit
| Feature | Synthetic Dyes | Genetically Encoded Calcium Indicators (GECIs) |
|---|---|---|
| Core Identity | Small, chemical molecules (e.g., BAPTA-based) [38] | Recombinant protein chimeras (e.g., calmodulin + GFP) [40] [42] |
| Introduction Method | Bulk loading via acetoxymethyl (AM) esters [44] [12] | Genetic delivery (transgenic animals, viral vectors) [12] [40] |
| Cell-Type Specificity | Limited; typically labels all cells in the loaded area [12] | High; enabled by cell-type-specific promoters or Cre-lines [12] [40] |
| Longitudinal Imaging | Short-term (hours); dye leaks and is metabolized [12] | Excellent; stable expression allows imaging over days to weeks [12] [42] |
| Subcellular Targeting | Difficult, primarily cytosolic | Straightforward; can be engineered for organelles (e.g., ER/SR with NEMOer) [43] |
| Invasiveness | Moderate; AM ester hydrolysis can buffer calcium and affect physiology [12] [40] | Variable; high expression of GECIs may chelate calcium and interfere with signaling [40] |
| Ease of Use | Simple, rapid protocol; no genetic manipulation needed [39] | Complex; requires molecular biology and/or animal work |
| Cost & Reagents | Recurring cost for dyes | Primarily initial setup cost (vectors, viruses) |
Table 2: Key Performance Metrics and Representative Indicators
| Metric | Synthetic Dyes | GECIs |
|---|---|---|
| Affinity (Kd) | Tunable (e.g., Fura-2: ~145 nM, NIR dyes: various) [38] | Tunable (e.g., GCaMP6s: high, NEMOer-f: ~µM-mM for ER) [42] [43] |
| Dynamic Range (ΔF/F) | Generally high (e.g., Cal-520: >6 for single APs [44]; NIR dyes: up to 1000-fold [38]) | Continuously improving (e.g., FR-GECO1c: 18-fold; NEMOer: up to 349-fold in cellulo) [33] [43] |
| Temporal Resolution | Fast (can detect single action potentials) [44] | Good to fast; latest variants (e.g., NEMOer-f, GCaMP8) detect single APs [41] [43] |
| Spectral Variety | Broad, from UV to NIR (Fura-2, Indo-1, Fluo-3/4, Cal-520, BioTracker NIR) [44] [38] [39] | Expanding (GCaMP-green, R-GECO-red, FR-GECO-far-red) [41] [33] |
| Representative Indicators | Fura-2 (rationetric), Fluo-4 (intensiometric), Cal-520 (high SNR), BioTracker NIR dyes [44] [38] [39] | GCaMP6s/f/m (green), jRGECO1a (red), FR-GECO1a/c (far-red), NEMOer (ER-targeted) [41] [33] [42] |
Decision Framework and Experimental Workflows
Choosing Your Tool: A Strategic Workflow
The following diagram outlines a systematic decision-making process for selecting between synthetic dyes and GECIs.
Detailed Experimental Protocols
Protocol for Synthetic Dye Imaging (exemplified with Fura-2 AM)
This protocol is adapted from established methods for measuring intracellular calcium in live cells [39].
Research Reagent Solutions:
- Fura-2 AM: The cell-permeable, esterified form of the ratiometric calcium indicator [39].
- HBSS (Hank's Buffered Salt Solution): A physiological buffer to maintain cell health during imaging [39].
- Pluronic F-127: A non-ionic surfactant used to disperse AM-ester dyes and prevent crystallization [44].
- BSA (Bovine Serum Albumin): Added to the loading solution to reduce dye sequestration and improve loading consistency [39].
Procedure:
- Reagent Preparation: Reconstitute lyophilized Fura-2 AM in high-quality DMSO to create a stock solution. Prepare working dye solution by diluting the stock in HBSS containing 1 mg/mL fatty-acid-free BSA and 0.02-0.05% Pluronic F-127. Sonicate and filter the solution to ensure dye dispersal [39].
- Cell Loading: Plate cells onto glass coverslips. On the day of imaging, wash cells with pre-warmed HBSS. Incubate cells with the Fura-2 AM working solution for 30-60 minutes at 37°C in the dark [39].
- Dye Desterification: Remove the dye solution and wash cells 3-4 times with fresh HBSS. Incubate for an additional 20-45 minutes to allow complete hydrolysis of the AM esters by intracellular esterases, activating the dye [39].
- Image Acquisition: Place the coverslip in a recording chamber on the microscope. Continuously perfuse with HBSS. Use a fluorescence microscope capable of rapid excitation switching. Collect emission at ~510 nm while alternately exciting at 340 nm and 380 nm. The ratio of fluorescence (F340/F380) is proportional to the intracellular calcium concentration [39].
- Stimulation & Controls: During imaging, stimulate cells with relevant agonists, receptor ligands, or high-K⁺ solution to depolarize membranes. Ionomycin, a calcium ionophore, can be used as a positive control to achieve maximum calcium elevation [39] [42].
Protocol for GECI Imaging (exemplified with GCaMP6s)
This protocol outlines the use of stably expressed GECIs for assays, including high-throughput screening [42].
Research Reagent Solutions:
- GCaMP6s-P2A-Bsr Plasmid: An expression vector where the GCaMP6s sequence is coupled to a blasticidin resistance gene via a self-cleaving P2A peptide, ensuring high and stable expression [42].
- Blasticidin S: An antibiotic used for the selection and maintenance of stably expressing cell lines.
- Appropriate Cell Culture Media: For the cell line of choice (e.g., HEK293, HeLa).
- Ionomycin: A calcium ionophore used for positive control stimulation [42].
Procedure:
- Cell Line Generation: Transfect the target cell line (e.g., HEK293) with the GCaMP6s-P2A-Bsr plasmid. Select stable clones by culturing in media containing Blasticidin S. The P2A linkage ensures that cells expressing the antibiotic resistance also express GCaMP6s, maintaining population homogeneity [42].
- Validation & Culturing: Validate GCaMP6s expression and function via fluorescence microscopy or flow cytometry. Stimulate with ionomycin to confirm a robust fluorescence increase. Maintain the validated clonal line under Blasticidin selection [42].
- Assay Setup: Plate the stable cells into the wells of a microtiter plate (96-, 384-, or 1536-well format). For GPCR or ion channel studies, cells may be co-transfected or stably express the target receptor/channel [42].
- High-Throughput Imaging: Use a fluorescent plate reader or high-content imager with standard FITC/GFP filter sets (Ex/~488 nm, Em/~510 nm). Add compound libraries or ligands and monitor fluorescence intensity (ΔF/F) in real-time. The stable expression eliminates the need for dye loading on the day of the assay, streamlining the process [42].
- Data Analysis: Analyze the kinetic fluorescence traces to identify compounds that modulate calcium flux through the target of interest.
Advanced Applications and Future Directions
The choice of indicator continues to enable new frontiers in research. Synthetic dyes like Cal-520, with their high signal-to-noise ratio, are indispensable for detecting single action potentials in vivo [44]. GECIs, with their genetic encodability, are the foundation of systems neuroscience, allowing researchers to map neural circuits and correlate the activity of thousands of neurons with behavior [41] [40]. They are also pivotal for studying specific cell types in complex tissues, such as somatosensory neurons in the dorsal root ganglion [12].
Future developments are focused on overcoming current limitations. Emerging GECIs are pushing further into the red and far-red spectrum (e.g., FR-GECOs) for deeper tissue penetration and reduced phototoxicity [33]. New engineering efforts are creating highly sensitive and dynamic indicators for organelles like the endoplasmic reticulum (e.g., NEMOer), enabling the study of subcellular calcium signaling events that were previously inaccessible [43]. Furthermore, the development of indicators with calcium-sensitive fluorescence lifetimes (FLIM) promises more quantitative measurements of absolute calcium concentration, independent of probe concentration or excitation light path length [45]. The ongoing refinement of both synthetic dyes and GECIs will undoubtedly continue to expand the boundaries of live-cell calcium imaging.
The accurate detection of calcium signaling in live cells is a cornerstone of modern physiological research, enabling scientists to decipher a universal language of cellular communication. Calcium ions (Ca²⁺) act as a multifaceted signaling molecule, regulating a vast array of physiological and developmental pathways in both plants and animals [46]. The transient and spatio-temporal variations in Ca²⁺ concentrations in response to stimuli, known as "Ca²⁺ signatures," are decoded by the cell to produce specific adaptive responses [46]. The study of these dynamics relies fundamentally on robust methods for introducing Ca²⁺ sensors into cells. This guide provides an in-depth technical examination of the three primary techniques for sensor loading and delivery—electroporation, AM esters, and viral transduction—framed within the context of live-cell calcium signaling research. The choice of delivery method directly impacts experimental outcomes, including the cell types that can be studied, the duration of expression, and the fidelity of the recorded Ca²⁺ transients.
Core Delivery Technologies
Electroporation
Mechanism and Workflow: Electroporation is a physical method that utilizes high-voltage electrical pulses to create transient, nanoscale pores in the cell membrane, through which nucleic acids or proteins can enter the cell [47]. This process involves suspending cells in an electroporation buffer containing the genetic material of interest (e.g., plasmid DNA encoding a Genetically Encoded Calcium Indicator or GECI) and applying an external electric field. The precise control over parameters such as pulse voltage, duration, and number is critical for maximizing uptake while maintaining cell viability [47].
Applications in Calcium Imaging: Electroporation is particularly effective for the transfection of hard-to-transfect primary cells, stem cells, and neurons. It allows for both transient and stable transfection and is adaptable for in vivo applications, such as in utero electroporation in model organisms. A key advantage for calcium imaging is the potential for co-transfection of multiple plasmids, enabling, for instance, the simultaneous expression of a GCaMP sensor and an optogenetic actuator to create an all-optical electrophysiology setup.
Limitations: A significant drawback is the frequent induction of high rates of cell death due to membrane damage [47]. The technique requires extensive, cell-type-specific optimization of electrical parameters and often results in variable transfection efficiency. Furthermore, the equipment can be costly and may not be amenable to all experimental formats, such as high-throughput screens.
Acetoxymethyl (AM) Esters
Mechanism and Workflow: AM esters represent a chemical method for delivering small molecule Ca²⁺ dyes (e.g., Fura-2, Indo-1) into cells. These dyes are chemically modified with acetoxymethyl ester groups, rendering them uncharged and lipophilic. This allows the dyes to passively diffuse across the cell membrane. Once inside the cytoplasm, ubiquitous intracellular esterases cleave the AM esters, releasing the charged, active form of the dye which is now trapped within the cell.
Applications in Calcium Imaging: AM ester dyes are the gold standard for rapid population-level loading of cells, including primary tissues and acutely isolated cells like neurons in brain slices. They are ideal for short-term experiments measuring Ca²⁺ dynamics in bulk cell populations or in single cells. Their simplicity of use—often involving just an incubation period—makes them accessible for labs without specialized equipment.
Limitations: This method is restricted to small molecule dyes and cannot be used for genetically encoded sensors like GCaMP. Dyes can be unevenly distributed within cells, potentially sequestering into organelles, and are susceptible to leakage or extrusion from the cytoplasm over time. Furthermore, the esterase activity required for dye activation can vary between cell types, and the cleavage byproducts may have unintended toxic effects on cellular physiology.
Viral Transduction
Mechanism and Workflow: Viral transduction employs engineered viruses, most commonly Adeno-Associated Viruses (AAVs) or lentiviruses, to deliver genetic material encoding GECIs into target cells. The process involves incubating cells or tissues with a preparation of viral particles containing the transgene of interest. The virus binds to specific cell surface receptors, is internalized, and its genetic payload is delivered to the nucleus to drive sustained expression of the sensor.
Applications in Calcium Imaging: Viral transduction is unparalleled for achieving long-term, stable, and cell-type-specific expression of GECIs in complex tissues and live animals [48] [49]. The use of cell-type-specific promoters (e.g., synapsin for neurons) allows for targeted expression. Advanced application methods, such as intracerebral injections for local delivery or systemic retro-orbital injections of novel serotypes like PHP.eB for brain-wide expression in mice, have vastly expanded its utility [49]. It is the method of choice for longitudinal studies and for introducing complex genetic tools like CaMPARI2, which permanently marks activated cells [48].
Limitations: Biosafety concerns and stringent regulatory requirements are major considerations. There is a limited cargo capacity for some viral vectors (especially AAVs), and the potential for immunogenicity can preclude repeated administration. Production of high-titer, pure viral stocks is complex and time-consuming. A critical technical challenge is optimizing transduction efficiency, which can be enhanced in tissue slices through methods like media agitation, physiological temperature culture, and removal of protease inhibitors [48].
Table 1: Comparative Analysis of Sensor Loading and Delivery Methods
| Feature | Electroporation | AM Esters | Viral Transduction |
|---|---|---|---|
| Primary Mechanism | Physical pore formation via electrical pulses [47] | Chemical; passive diffusion & enzymatic activation | Biological; viral infection & transgene expression [48] [49] |
| Typical Cargo | DNA, RNA, proteins | Small molecule Ca²⁺ dyes (e.g., Fura-2, Indo-1) | DNA encoding GECIs (e.g., GCaMP, jRGECO) [48] [49] |
| Expression Duration | Transient to stable (if integrated) | Short-term (hours) | Long-term (weeks to months) [49] |
| Key Advantages | Effective in hard-to-transfect cells; parameter control [47] | Rapid, simple, no genetic modification | High efficiency; cell-type specificity; suitable for in vivo use [48] [49] |
| Key Limitations | High cell toxicity; requires optimization [47] | Dye compartmentalization/leakage; not for GECIs | Immunogenicity; limited cargo; complex production [47] |
| Ideal Use Case | Single-cell electroporation in slices; primary cell transfection | Acute brain slice imaging; population kinetics | Long-term in vivo imaging; targeted expression [49] |
Advanced Technical Considerations
Quantitative Method Selection
Choosing the appropriate delivery method requires a quantitative understanding of its performance characteristics. The following table summarizes key metrics to guide researchers in selecting a technique aligned with their experimental goals, from high-throughput screening to chronic in vivo imaging.
Table 2: Quantitative Performance Metrics of Delivery Methods
| Metric | Electroporation | AM Esters | Viral Transduction (AAV) |
|---|---|---|---|
| Typical Efficiency | Variable (10-80%), highly cell-type dependent [47] | Very high (>90% of cells in a population) | High (can approach >90% in vitro) [48] |
| Time to Experiment | 24-72 hours (for DNA expression) | 30 minutes - 2 hours | 1-4 weeks for full expression [49] |
| Cell Viability Post-Delivery | Often low; requires significant optimization [47] | High, though esterase byproducts can be toxic | Generally high, dependent on serotype and MOI |
| Spatial Targeting Precision | High (can be applied to single cells in situ) | Low (bulk loading of cell populations) | High with targeted injection; cell-type specific with promoters [49] |
| Throughput Potential | Medium (96-well systems available) | Very High (simple plate-based incubation) | Low to Medium (depends on production scale) |
Detailed Experimental Protocol: Adenoviral Transduction in Pancreas Tissue Slices
The following detailed protocol, adapted from a recent preprint on transducing human pancreas tissue slices with the CaMPARI2 biosensor, exemplifies the optimization required for effective viral delivery in complex ex vivo systems [48].
1. Tissue Slice Preparation:
- Generate 120 µm thick live pancreatic tissue slices using a vibratome, following established protocols [48].
- Allow slices to recover for 24 hours in an incubator set to 24°C and 5% CO₂ in slice culture media (e.g., Dulbecco’s Low Glucose Modified Eagles Medium supplemented with 10% FBS and 25 kIU/ml aprotinin) [48].
2. Viral Transduction:
- Transfer slices to 35 mm dishes (2 slices per dish) with 2 ml of slice culture media without protease inhibitor (e.g., aprotinin). Critical Note: Removal of the protease inhibitor significantly enhances viral transduction efficiency [48].
- Add the adenoviral vector (e.g., 2.6 × 10⁸ PFU per tissue slice) directly to the dish [48].
- Culture the slices on an orbital shaker set to 40 rpm inside a 37°C incubator for 24 hours. Agitation is a key factor for improving viral penetration throughout the slice volume [48].
- After 24 hours, replace the virus-containing media with fresh culture media (without protease inhibitor) and culture for an additional 24 hours on the orbital shaker at 37°C before imaging or functional assays [48].
3. Validation and Functional Assay:
- Confirm tissue viability and function through glucose-stimulated insulin secretion perifusion assays [48].
- For CaMPARI2, expose transduced slices to photoconverting light during high-glucose stimulation to permanently mark active cells, then fix and immunostain for correlation with spatial and morphological data [48].
The Scientist's Toolkit: Research Reagent Solutions
Successful execution of delivery protocols relies on a suite of specialized reagents and tools. The following table catalogues essential materials for the featured viral transduction protocol and the broader field.
Table 3: Essential Research Reagents and Materials
| Reagent / Material | Function / Application | Example Use Case |
|---|---|---|
| Adeno-Associated Virus (AAV) | Efficient in vivo and in vitro gene delivery with low immunogenicity [49] | Brain-wide expression of jGCaMP8s in mice via retro-orbital injection [49] |
| Lipid Nanoparticles (LNPs) | Non-viral encapsulation and delivery of nucleic acids (mRNA, siRNA) [47] | Delivery of mRNA-based therapeutics and vaccines; emerging for research tool delivery. |
| Poly(β-amino ester)s | Biodegradable cationic polymers for nucleic acid delivery; tunable via end-modification [50] | High-throughput screening of polymer variants to optimize gene delivery efficiency [50]. |
| GCaMP8s/m/f | Genetically Encoded Calcium Indicators (GECIs) with high sensitivity and fast kinetics [49] | In vivo two-photon calcium imaging of neural activity in the primary visual cortex [49]. |
| CaMPARI2 | Photoconvertible calcium integrator that permanently marks active cells [48] | High-throughput mapping of glucose-induced calcium activity in all islets within a pancreas slice [48]. |
| GreenT-ECs | Ultra-low affinity GECIs for imaging interstitial (extracellular) calcium [51] | Monitoring homeostatic regulation of tissue interstitial calcium in transgenic zebrafish and rodent hippocampus [51]. |
Schematic Workflows
The following diagrams illustrate the logical workflow and key decision points for selecting and implementing a sensor delivery method, and the specific procedural steps for viral transduction in tissue slices.
Diagram 1: Sensor Delivery Method Selection
Diagram 2: Viral Transduction Workflow
Electroporation, AM esters, and viral transduction form a complementary toolkit for loading calcium sensors into live cells, each with distinct mechanistic principles and application landscapes. The choice of method is not one-size-fits-all but must be strategically aligned with the experimental question, considering the sensor type, target cells, and required expression dynamics. As the field advances, the development of novel delivery technologies—such as engineered virus-like particles (VLPs) for editor delivery [52] and improved non-viral systems [47]—alongside next-generation sensors with targeted localization [49] and tailored affinities [51] will continue to push the boundaries of our ability to visualize and interpret the intricate dynamics of calcium signaling in health and disease.
The study of dynamic biological processes, such as calcium signaling in live cells, relies heavily on the ability to visualize these events with high spatial and temporal fidelity. The choice of microscopy modality is paramount, as it directly impacts the resolution, depth, and viability of long-term imaging. Widefield, confocal, and two-photon microscopy represent three cornerstone techniques for fluorescence imaging, each with distinct principles, capabilities, and optimal application ranges. Framed within the context of calcium signaling detection, this guide provides an in-depth technical comparison of these modalities. It details their fundamental operating principles, presents a structured quantitative comparison, outlines specific experimental protocols for live-cell calcium imaging, and visualizes the core concepts to inform researchers and drug development professionals in selecting the most appropriate tool for their investigative needs.
Core Principles and Technical Comparison
Fundamental Operating Principles
Widefield Fluorescence Microscopy: In this technique, the entire specimen is illuminated with light of a specific excitation wavelength [53]. The resulting fluorescence from both in-focus and out-of-focus planes is collected by the detector, often leading to a blurred image with high background signal. This lack of optical sectioning is its primary limitation for imaging thick samples [54] [55].
Confocal Laser Scanning Microscopy (CLSM): Confocal microscopy achieves optical sectioning by using a focused laser beam to scan the sample point-by-point and a pinhole aperture placed in front of the detector to block out-of-focus light [56] [54] [55]. This process results in a sharp image with significantly reduced background haze, allowing for the collection of clear optical sections that can be reconstructed into 3D models [55] [57]. Its ability to eliminate out-of-focus light is its key advantage over widefield microscopy [54].
Two-Photon Excitation (TPE) Microscopy: Two-photon microscopy also employs a focused laser beam for scanning but relies on the near-simultaneous absorption of two long-wavelength (typically infrared) photons to excite a fluorophore [56] [58]. This excitation only occurs at the focal point where the photon density is highest, providing inherent optical sectioning without the need for a detection pinhole [58]. The use of longer wavelengths reduces light scattering and allows for deeper penetration into biological tissues, making it the gold standard for intravital and deep-tissue imaging [56] [58].
Quantitative Technical Comparison
The table below summarizes the key characteristics of each microscopy modality, with a specific focus on parameters critical for calcium signaling research in live cells.
Table 1: Technical Comparison of Widefield, Confocal, and Two-Photon Microscopy for Live-Cell Imaging
| Feature | Widefield Microscopy | Confocal Microscopy | Two-Photon Microscopy |
|---|---|---|---|
| Optical Sectioning | No | Yes, via pinhole [54] [55] | Yes, inherent via non-linear excitation [58] |
| Excitation Volume | Full sample volume | Diffraction-limited spot | Diffraction-limited spot |
| Typical Excitation Wavelength | UV-Visible | UV-Visible [54] | Infrared (~2x one-photon wavelength) [56] [58] |
| Penetration Depth | Limited (up to ~10s of µm) | Good for mildly scattering samples (up to ~200 µm) [56] | Excellent for scattering samples (up to millimeters) [56] [58] |
| Lateral Resolution | ~200-300 nm | ~200-250 nm [56] | ~200-400 nm (wavelength-dependent) |
| Axial Resolution | Low | ~500-700 nm [55] | ~1-2 µm |
| Background Signal | High | Low [55] | Very low in deep tissue |
| Live-Cell Viability | Moderate (risk of phototoxicity from full-volume illumination) | Lower (phototoxicity and photobleaching from focused laser) [55] [59] | Higher (reduced out-of-focus photobleaching & phototoxicity) [58] |
| Relative Speed | Fast (camera-based) | Slower (point-scanning) [55] [59] | Slow (point-scanning) but faster resonant scanners available [58] |
| Relative Cost | Low | High [55] [59] | Very High [56] [58] |
Experimental Protocols for Calcium Signaling Detection
Calcium signaling is a rapid and dynamic process. The following protocols are generalized for capturing these events using genetically encoded calcium indicators (GECIs) like GCaMP in live cells or tissues.
Protocol 1: Rapid Calcium Transient Imaging with Widefield Microscopy and Deconvolution
This protocol is optimized for speed and accessibility, achieving subnuclear axial resolution in tissues up to 500 µm deep when combined with deconvolution [60].
- Sample Preparation: Transfer adherent cells expressing a GECI (e.g., GCaMP6s) to an imaging chamber. For tissues, perform clearing using methods like ADAPT-3D or CUBIC to achieve transparency and stain with a nuclear marker if needed [60].
- Microscope Configuration: Use a widefield epifluorescence microscope equipped with:
- A high-NA objective (e.g., NA 1.0) with a long working distance and a correction collar for spherical aberrations [60].
- A precise electronically controlled z-drive.
- An LED light source and a sensitive sCMOS or CMOS camera.
- Appropriate excitation/emission filters for the GECI.
- Image Acquisition:
- Post-Processing (Deconvolution):
- Use deconvolution software (e.g., Scientific Volume Imaging Huygens, DeconvolutionLab2) with a theoretically derived, depth-variant Point Spread Function (PSF) [60].
- Input parameters: lens immersion refractive index, tissue embedding refractive index, and distance from the coverslip to the tissue start [60].
- Process the z-stack to computationally remove out-of-focus light and restore a sharp, optically sectioned image.
Protocol 2: High-Resolution 3D Calcium Imaging in Live Cells with Confocal Microscopy
This protocol prioritizes high-contrast, optical sectioning for 3D reconstruction of calcium signals in cell cultures or thin tissue slices.
- Sample Preparation: Culture cells expressing GCaMP on glass-bottom dishes. For tissues, prepare 100-200 µm thick acute slices.
- Microscope Configuration: Use a laser scanning confocal microscope with:
- A high-resolution oil or water immersion objective (e.g., 60x, NA 1.4).
- Laser lines matched to the GECI excitation (e.g., 488 nm for GCaMP).
- The confocal pinhole set to 1 Airy unit for optimal sectioning.
- Photomultiplier tubes (PMTs) or hybrid detectors (e.g., GaAsP).
- Image Acquisition:
- Set the scan speed appropriately; use resonant scanners for faster events.
- Define a z-stack range with step sizes of 0.5-1 µm.
- Keep laser power as low as possible to minimize photobleaching and phototoxicity.
- For time-lapse 3D imaging, the interval between z-stacks will be limited by the acquisition speed.
Protocol 3: Deep-Tissue Calcium Imaging in Brain Slices or In Vivo with Two-Photon Microscopy
This protocol is designed for imaging calcium dynamics deep within scattering tissues, such as in brain slices or in live animals.
- Sample Preparation: For in vivo imaging, use a cranial window in an anesthetized or head-fixed behaving animal expressing GCaMP in specific neuronal populations. For ex vivo work, use thick brain slices (300-500 µm).
- Microscope Configuration: Use a two-photon microscope with:
- A tunable, mode-locked Ti:Sapphire laser for excitation (e.g., tuned to ~920 nm for GCaMP).
- A high-NA, long-working-distance water immersion objective.
- Non-descanned detectors (NDDs) placed close to the objective to maximize collection of scattered emission photons [58].
- Image Acquisition:
- Locate the region of interest. The microscope can image hundreds of micrometers deep, even through the intact dura [58].
- Adjust the laser power to sufficient levels for excitation while monitoring for signs of tissue damage.
- Acquire images at a frame rate sufficient to capture calcium transients (often 5-30 Hz). The system can be combined with modules like Ex2p for imaging in curved tissues or behavioral setups [61].
The Scientist's Toolkit: Essential Reagents and Materials
Table 2: Key Research Reagent Solutions for Live-Cell Calcium Imaging
| Item Name | Function/Description | Example Use Case |
|---|---|---|
| Genetically Encoded Calcium Indicators (GECIs) | Fluorescent proteins (e.g., GCaMP6f, jGCaMP7s) whose brightness changes upon binding Ca²⁺. Provide genetic targeting to specific cell types. | Expression in neuronal populations via viral vectors or transgenics for in vivo calcium imaging [61]. |
| Synthetic Calcium Dyes | Small molecule dyes (e.g., Fluo-4 AM, Fura-2 AM). Cell-permeable AM esters facilitate loading into live cells. | Bulk-loading of cell populations in cultured cells or acute tissue slices where genetic manipulation is not feasible. |
| Tissue Clearing Reagents | Chemical mixtures (e.g., CUBIC, DISCO, ADAPT-3D) that render tissues transparent by refractive index matching [60]. | Enabling deep imaging (up to 500 µm) in fixed tissues with widefield microscopy [60]. |
| Mounting Media (Refractive Index Matched) | Specimen immersion media with a refractive index matching the objective lens and tissue (e.g., for oil, glycerol, or water immersion). | Crucial for minimizing spherical aberration and maintaining resolution at depth in all modalities [60] [55]. |
| Deconvolution Software | Computational software (e.g., Huygens, DeconvolutionLab2) that uses a Point Spread Function (PSF) to remove out-of-focus light [60]. | Restoring sharpness and achieving optical sectioning from widefield z-stacks, enabling high-resolution deep imaging [60]. |
Visualization of Core Concepts
Light Path and Signal Detection Principles
The following diagram illustrates the fundamental differences in how each microscopy modality illuminates the sample and collects the resulting fluorescence signal, which is the origin of their distinct performance characteristics.
Decision Workflow for Calcium Imaging Modality Selection
This flowchart provides a structured approach to selecting the most suitable microscopy technique based on key experimental parameters.
Calcium ions (Ca²⁺) serve as a ubiquitous intracellular messenger, regulating processes from neuronal excitation to gene transcription [18]. Understanding the spatial and temporal dynamics of these signals in living organisms is therefore crucial for advancing research in physiology, neuroscience, and drug development. In vivo calcium imaging has emerged as a powerful technique for this purpose, allowing scientists to monitor cellular activity in real-time within intact, functioning biological systems. Among the various methodologies developed, fiber photometry and microendoscopy have become cornerstone techniques, each offering unique advantages for monitoring calcium dynamics in deep brain structures or in freely behaving animals. This technical guide provides an in-depth examination of these core techniques, their associated reagents, and their application in chronic recording paradigms, framing them within the broader context of calcium signaling detection in live cell research.
Core Principles of Calcium Imaging In Vivo
In vivo calcium imaging relies on detecting transient increases in intracellular calcium concentration, which serve as a reliable proxy for cellular activity, particularly neuronal firing [62] [18]. When a neuron fires an action potential, voltage-gated calcium channels open, leading to a rapid influx of calcium ions. This flux can be captured and measured using calcium-sensitive indicators that change their fluorescent properties upon binding Ca²⁺.
The functional architecture of a calcium imaging experiment can be visualized as a sequential pathway from the biological event to the recorded signal. The following workflow outlines this fundamental process:
This cascade converts a biological event into a quantifiable optical signal. Two primary classes of indicators are used for this purpose: chemical indicators (e.g., Fura-2, Fluo-4) and genetically encoded calcium indicators (GECIs) like the GCaMP series [62] [35]. GECIs, which are engineered fluorescent proteins, have gained prominence for in vivo applications due to their ability for genetic targeting to specific cell types and stable long-term expression [62] [63].
Method 1: Fiber Photometry
Fiber photometry is an optical technique that enables the recording of bulk neural activity in freely moving animals [62] [63]. Its fundamental principle involves transmitting excitation light of a specific wavelength through an implanted optical fiber to excite fluorescent calcium indicators in the surrounding brain tissue (typically within a 50-400 μm radius). The emitted fluorescence is then collected back through the same fiber and delivered to a photodetector [62]. The resulting digitized light intensity signal reflects the relative concentration of calcium-bound sensors at the fiber tip, providing a "global" readout of population-level activity in a defined brain region [62].
The experimental setup and signal flow in a fiber photometry system can be summarized as follows:
A key advantage of fiber photometry is its minimal invasiveness compared to other techniques, coupled with its compatibility with complex behavioral paradigms [63]. Furthermore, its data dimensionality is relatively low compared to electrophysiology or two-photon imaging, simplifying analysis and sharing [62].
Experimental Protocol
Key Materials:
- Fiber photometry system: Comprising a light source (LED or laser), fluorescence detector, and optical components (dichroic mirrors, filters).
- Optic fiber implant: A low-autofluorescence optical fiber (200-400 μm diameter) housed in a ferrule.
- Calcium indicator: Typically a GECI such as GCaMP, delivered via viral vector (e.g., AAV) or expressed in transgenic animals.
- Stereotaxic surgery equipment: For precise implantation of the optic fiber.
Procedure:
Indicator Expression:
- Inject a viral vector (e.g., AAV-syn-GCaMP) into the target brain region of an anesthetized animal using stereotaxic coordinates. Allow 2-4 weeks for sufficient expression.
Fiber Implantation:
- Following indicator expression, implant the optic fiber ferrule above the target region. Secure the ferrule to the skull using dental cement.
Data Acquisition:
- After recovery, connect the animal's implant to the photometry system via a patch cord.
- Deliver excitation light (e.g., ~470 nm for GCaMP) and record the emitted fluorescence (e.g., ~510 nm for GCaMP) synchronously with behavioral data.
- Record the fluorescence signal, often alongside an isosbestic control signal (e.g., ~405 nm), which is calcium-insensitive and controls for motion artifacts and photobleaching [64].
Data Analysis:
- Process the raw fluorescence signal (F) to calculate ΔF/F, which represents the change in fluorescence relative to the baseline.
- The formula is: ΔF/F = (F - F₀) / F₀, where F₀ is the baseline fluorescence, often derived from the isosbestic control channel or a fitted exponential curve [64].
- Align calcium transients with behavioral event timestamps to correlate neural activity with specific actions or stimuli.
Method 2: Microendoscopy
Microendoscopy extends the reach of conventional microscopes into deep brain structures using miniature optical probes, primarily gradient refractive index (GRIN) lenses [65]. These needle-like lenses are inserted into the brain, where they relay images from deep tissue layers to the surface of the fiber, where they can be captured by a microscope objective or a miniaturized "miniscope" mounted on an animal's head [65] [66]. Unlike fiber photometry, which provides a bulk fluorescence signal, microendoscopy enables cellular-resolution imaging of individual neurons and their calcium dynamics in freely behaving animals [65].
The key advantage of GRIN lenses is their ability to overcome the penetration depth limits (~500-700 μm) of conventional two-photon microscopy, allowing access to subcortical structures like the hippocampus or hypothalamus [65]. Microendoscopes are versatile and compatible with various contrast modalities, including epifluorescence, two-photon excited fluorescence, and second-harmonic generation [65].
Table 1: Optical Parameters of Common Microendoscope Probes
| Microendoscope Type | Diameter (mm) | Length (mm) | Field of View (μm) | Lateral Resolution (μm) | Numerical Aperture (NA) |
|---|---|---|---|---|---|
| Singlet GRIN (0.46 pitch) | 1.0 | 4.4 | 700 | 0.9 | 0.49 |
| Singlet GRIN (0.94 pitch) | 0.5 | 4.3 | 350 | 1.0 | 0.47 |
| GRIN/plano-convex doublet (BK7 lens) | 1.0 | 3.7 | 120 | 0.8 | 0.65 |
| GRIN/plano-convex doublet (LaSFN9 lens) | 1.0 | 4.0 | 75 | 0.6 | 0.82 |
Data adapted from [65]. FWHM resolution is given for two-photon imaging at 920 nm.
Experimental Protocol
Key Materials:
- GRIN lens probe: Selected based on desired field of view, resolution, and target depth (see Table 1).
- Miniscope or microscope: A conventional upright microscope for anesthetized preparations, or a portable miniaturized microscope (miniscope) for freely moving behavior.
- Calcium indicator: Synthetic dyes (e.g., OGB-1) or GECIs delivered via viral injection.
Procedure:
Lens Implantation:
- Inject a calcium indicator into the deep brain region of interest.
- Implant a GRIN lens above the injected area, ensuring its focal plane is centered on the region of interest. Secure the lens to the skull.
Image Acquisition:
- For freely moving behavior, attach a compact miniscope to the GRIN lens implant.
- Acquire video-rate fluorescence image sequences of the brain region over time.
Data Processing and Analysis:
- Correct the raw video data for motion artifacts.
- Perform image segmentation to identify individual neurons as Regions of Interest (ROIs).
- Extract fluorescence time series (F(t)) for each ROI.
- Calculate ΔF/F for each cell to identify calcium transients, which correspond to neuronal activation.
Chronic Calcium Recordings
Long-term calcium imaging over days, weeks, or even months is highly desirable for studying chronic processes like learning, memory, neural development, and disease progression [67]. However, this presents specific challenges, primarily the cytotoxicity associated with long-term, high-level expression of GECIs like GCaMP [67].
The conventional GCaMP probe is based on calmodulin (CaM) and can interfere with endogenous CaM-binding proteins and signaling pathways, leading to nuclear accumulation of the probe, aberrant calcium oscillations, and impaired neurite outgrowth [67]. To address this, GCaMP-X was engineered by appending an apoCaM-binding motif and localization tags. This design sequesters the probe's CaM domain, preventing it from interfering with native signaling and significantly improving neuronal health during chronic expression [67].
The experimental design for a chronic imaging study must carefully consider the choice of indicator and expression system, as outlined below:
For chronic studies, it is critical to use reduced viral titers or transgenic animals with conditionally expressed indicators to minimize cytotoxicity. GCaMP-X provides a superior solution for such longitudinal experiments, enabling stable imaging over periods of one month or longer with significantly less damage to neuronal morphology and function [67].
The Scientist's Toolkit
Successful execution of in vivo calcium imaging requires a suite of specialized reagents and tools. The following table catalogues essential solutions for setting up these experiments.
Table 2: Research Reagent Solutions for In Vivo Calcium Imaging
| Item | Function | Examples & Key Characteristics |
|---|---|---|
| Genetically Encoded Calcium Indicators (GECIs) | Fluorescent proteins that change intensity upon calcium binding; enable genetic targeting and long-term expression. | GCaMP6/7/8: High sensitivity, fast kinetics. GCaMP-X: Engineered for reduced cytotoxicity in chronic studies [67]. RCaMP: Red-shifted variant for multiplexing. |
| Chemical Calcium Indicators | Synthetic dyes that fluoresce upon calcium binding; often used for acute experiments. | OGB-1 (Oregon Green BAPTA-1): Often used with bulk loading. Fluo-4, Fura-2: Ratiometric or intensity-based dyes [18] [35]. |
| Viral Vector Systems | Deliver genes encoding GECIs to specific brain regions; enable cell-type-specific expression. | Adeno-Associated Virus (AAV): Low immunogenicity, long-term expression. Cre-dependent AAVs: For cell-type-specific expression in transgenic Cre lines [62]. |
| Optic Fibers & Implants | Deliver excitation light and collect emitted fluorescence in fiber photometry. | Low-autofluorescence fibers: 200-400 μm diameter, housed in a ceramic ferrule [62] [63]. |
| GRIN Lenses | Relay images from deep brain structures to the surface for microendoscopy. | Singlet GRIN: Large field of view. GRIN doublet: Higher resolution, smaller field of view (see Table 1) [65]. |
| Analysis Software | Process raw fluorescence data, extract signals, and correlate with behavior. | GuPPy (Python): Open-source tool for analyzing fiber photometry data [64]. Custom MATLAB scripts: For miniscope data analysis (e.g., CNMF-E). |
Fiber photometry and microendoscopy represent two powerful, complementary approaches for detecting calcium signals in vivo. The choice between them hinges on the specific research question. Fiber photometry is optimal for monitoring the aggregate activity of a defined neural population over extended periods with high temporal resolution and is highly compatible with complex behaviors. In contrast, microendoscopy is the method of choice when single-cell resolution within a deep brain circuit is required. For both techniques, the ongoing development of improved calcium indicators, such as the less cytotoxic GCaMP-X, is pushing the boundaries of chronic imaging, enabling longitudinal studies of neural plasticity and disease progression. As these technologies continue to mature and become more accessible, they will undoubtedly unlock deeper insights into the complex "calcium code" that underlies brain function and dysfunction, providing critical pathways for future therapeutic development.
Calcium ions (Ca²⁺) function as ubiquitous intracellular messengers, regulating processes from neurotransmitter release and muscle contraction to gene transcription and cell death [68]. The specificity of these diverse cellular responses is governed by sophisticated spatiotemporal patterns of Ca²⁺ concentrations within specialized compartments [69]. This technical guide details established and emerging methodologies for interrogating Ca²⁺ dynamics in three critical compartments: the cytosol, endoplasmic reticulum/sarcoplasmic reticulum (ER/SR), and mitochondria. We provide a comparative analysis of indicator tools, detailed experimental protocols, and data analysis frameworks, contextualized within live-cell research and drug development applications. Mastering these compartment-specific imaging techniques enables researchers to decipher the complex language of Ca²⁺ signaling in health and disease.
Principles of Calcium Compartmentalization
Intracellular Ca²⁺ signaling depends on maintaining steep concentration gradients across organelle membranes. In resting cells, cytosolic Ca²⁺ ([Ca²⁺]c) is maintained at ~100 nM, while the ER/SR lumen ([Ca²⁺]ER) reaches 0.2-2 mM, and mitochondrial matrix ([Ca²⁺]m) levels are similar to the cytosol [68]. Upon stimulation, [Ca²⁺]c can rapidly increase to 1-3 µM, triggering compartment-specific responses.
The cytosol serves as the primary conduit for Ca²⁺ signals, integrating inputs from plasma membrane channels and intracellular stores. The ER/SR represents the major intracellular Ca²⁺ store, equipped with Sarco/Endoplasmic Reticulum Ca²⁺ ATPases (SERCAs) for Ca²⁺ uptake, and inositol 1,4,5-trisphosphate receptors (IP₃Rs) or ryanodine receptors (RyRs) for controlled release [68]. Mitochondria temporarily sequester cytosolic Ca²⁺ via the Mitochondrial Calcium Uniporter (MCU) complex, using the organelle's negative membrane potential as a driving force. Mitochondrial Ca²⁺ efflux occurs through Na⁺/Ca²⁺ and H⁺/Ca²⁺ exchangers [68]. This dynamic cycling between compartments forms an integrated signaling network known as the ER mitochondria calcium cycle [70].
Table 1: Characteristic Calcium Concentrations and Regulatory Proteins in Cellular Compartments
| Compartment | Resting [Ca²⁺] | Stimulated [Ca²⁺] | Key Influx Mechanisms | Key Efflux/Sequestration Mechanisms |
|---|---|---|---|---|
| Cytosol | ~100 nM | 1-3 µM | IP₃R, RyR, Plasma membrane channels | PMCA, NCX, SERCA, MCU |
| ER/SR | 0.2-2 mM | Variable (depletes with release) | SERCA | IP₃R, RyR |
| Mitochondria | ~100 nM (can transiently reach µM range) | Can exceed 10 µM during overload | MCU Complex | mNCX, H⁺/Ca²⁺ exchanger |
Calcium Indicator Selection
Choosing appropriate calcium indicators is fundamental to successful compartment-specific imaging. Researchers must consider the chemical versus genetic encoding of indicators, targeting specificity, binding affinity (Kd), and photophysical properties.
Chemical Calcium Indicators
Chemical indicators are small molecules that bind Ca²⁺ via chelation, often using BAPTA-based structures for high Ca²⁺ specificity and pH stability [71]. These dyes are typically modified with acetoxymethyl esters (AM) that render them membrane-permeant. Once inside the cell, endogenous esterases cleave the AM groups, trapping the charged indicator intracellularly [71].
Table 2: Chemical Calcium Indicators for Compartment-Specific Imaging
| Indicator | Excitation/Emission | Binding Kd | Compartment | Key Features and Applications |
|---|---|---|---|---|
| Fura-2 AM | 340/380 nm ex, 510 nm em | ~145 nM | Cytosol | Ratiometric, UV-excitable, reduced photo-bleaching artifacts [72] [71] |
| Indo-1 AM | 361 nm ex, 405/485 nm em | ~230 nM | Cytosol | Ratiometric, flow cytometry applications [71] |
| Fluo-3 AM | 506 nm ex, 526 nm em | ~390 nM | Cytosol | Intensity-based, large signal increase upon Ca²⁺ binding [71] |
| Quin-2 AM | 339 nm ex, 492 nm em | ~60 nM | Cytosol | High Ca²⁺ affinity, suitable for low [Ca²⁺] monitoring [71] |
| Rhod-2 AM | 552 nm ex, 581 nm em | ~570 nM | Mitochondria | Positively charged, naturally accumulates in mitochondria [71] |
| BioTracker NIR Dyes | Near-infrared | Variable | Cytosol | Enable multicolor imaging with GFP/RFP reporters [71] |
Genetically Encoded Calcium Indicators (GECIs)
GECIs represent a transformative advancement for compartment-specific imaging, as they can be precisely targeted to subcellular locations using genetic targeting sequences [68]. Cameleon indicators are FRET-based sensors that undergo changes in fluorescence resonance energy transfer efficiency upon Ca²⁺ binding, providing rationetric measurements ideal for quantitative applications [68]. Other GECI families include single-fluorophore sensors like GCaMP, which combine circularly permuted GFP with calmodulin and M13 peptide domains.
Experimental Protocols
Cytosolic Calcium Imaging Using Chemical Indicators
Materials:
- Culture medium without serum or phenol red
- Appropriate calcium indicator (e.g., Fura-2-AM, Fluo-3-AM)
- Pluronic F-127 detergent (0.02% w/v)
- Dimethyl sulfoxide (DMSO)
- Hanks' Balanced Salt Solution (HBSS) or similar physiological buffer
- Cell culture vessel with cells of interest
- Fluorescence microscope with appropriate filters and temperature control
Procedure:
- Indicator Preparation: Prepare 1-5 mM stock solution of the AM-ester indicator in anhydrous DMSO. For difficult-to-load cells, add Pluronic F-127 to improve dispersion.
- Cell Loading: Replace culture medium with loading solution containing 1-10 µM indicator in serum-free medium or buffer. Incubate for 20-60 minutes at 20-37°C depending on cell type and indicator permeability.
- Ester Hydrolysis: Replace loading solution with fresh culture medium or buffer and incubate for additional 20-30 minutes to ensure complete de-esterification.
- Image Acquisition: Mount samples on microscope and acquire images using appropriate excitation/emission settings. For ratiometric imaging with Fura-2, alternate between 340 nm and 380 nm excitation while collecting emission at 510 nm.
- Stimulation: Apply pharmacological agents or physiological stimuli during continuous imaging to monitor dynamic Ca²⁺ responses.
- Calibration (for quantitative measurements): Perform in situ calibration using ionomycin (10 µM) in Ca²⁺-free buffer with EGTA (5 mM) for minimum ratio (Rmin), followed by saturating Ca²⁺ (10 mM) for maximum ratio (Rmax).
Mitochondrial and Cytosolic Imaging Using GECIs
Materials:
- Plasmid DNA encoding targeted cameleon indicator (e.g., mito-Cameleon for mitochondria, cyto-Cameleon for cytosol)
- Transfection reagent or viral delivery system
- Appropriate cell culture medium
- Fluorescence microscope with FRET capability or appropriate filter sets
- Image acquisition software capable of ratio imaging
Procedure:
- Sensor Expression: Transfert cells with plasmids encoding compartment-targeted cameleon indicators. For mitochondrial targeting, indicators typically include a mitochondrial localization sequence (e.g., from cytochrome c oxidase). For ER targeting, include an ER retention signal (e.g., KDEL sequence).
- Expression Validation: Allow 24-48 hours for expression. Verify proper subcellular localization using confocal microscopy.
- FRET Imaging: For cameleon imaging, sequentially excite CFP at 440 nm while collecting emission at both 480 nm (CFP channel) and 535 nm (FRET channel). The ratio of YFP/CFP emission provides a Ca²⁺-dependent signal independent of indicator concentration.
- Stimulation and Recording: Acquire baseline images before applying stimuli. For mitochondrial Ca²⁺ measurements, monitor responses to agonists that trigger ER Ca²⁺ release (e.g., ATP, histamine) or store-operated Ca²⁺ entry.
- Data Analysis: Calculate ratio values (YFP/CFP for cameleon) for each time point and normalize to baseline. Convert ratio values to [Ca²⁺] using established calibration curves specific to each indicator variant.
Calcium Imaging in 3D Organoid Systems
Materials:
- Intestinal or other tissue-derived organoids
- Fura-2-AM or other AM-ester indicator
- Advanced Dulbecco's Modification of Eagle Medium (DMEM)/F12
- Matrigel or other extracellular matrix substitute
- Confocal or two-photon microscope
Procedure:
- Organoid Culture: Maintain organoids in Matrigel droplets with appropriate growth factor-supplemented medium [72].
- Indicator Loading: Incubate organoids with 2-5 µM Fura-2-AM in medium for 45-90 minutes at 37°C [72].
- Washing and Recovery: Replace loading solution with fresh medium and incubate for 30 minutes to ensure complete de-esterification.
- Image Acquisition: Mount organoids on microscope and image using appropriate modality. Light-sheet microscopy provides excellent penetration for 3D structures while minimizing phototoxicity [72] [73].
- Analysis: Segment individual cells within organoids and extract fluorescence traces. Analyze Ca²⁺ oscillations in response to nutrients, drugs, or hormones to assess functional responses [72].
Data Processing and Analysis
Calcium imaging data requires specialized processing to extract meaningful biological information from often noisy signals.
Preprocessing Pipeline:
- Denoising: Apply algorithms like ND-SAFIR (non-local means denoising) or DeepInterpolation (self-supervised deep learning) to reduce Poisson-Gaussian noise inherent in fluorescence microscopy [73].
- Motion Correction: Use rigid or non-rigid registration algorithms (e.g., NoRMcorre) to correct for sample drift or movement, particularly important in live-cell and in vivo imaging [73].
- Background Subtraction: Subtract background fluorescence from regions without cells.
Quantitative Analysis:
- Region of Interest (ROI) Definition: Manually or automatically define ROIs corresponding to cellular compartments or entire cells.
- Trace Extraction: Calculate average fluorescence intensity within each ROI across all frames.
- Ratiometric Conversion: For ratiometric indicators, compute ratio values frame-by-frame.
- Delta F/F0 Calculation: For single-wavelength indicators, normalize traces as (F-F0)/F0, where F0 represents baseline fluorescence.
- Event Detection: Identify Ca²⁺ transients using threshold-based or template-matching algorithms.
- Statistical Analysis: Quantify amplitude, frequency, kinetics (rise time, decay time), and spatial spread of Ca²⁺ signals.
For compartment-specific analysis, simultaneously monitor multiple regions representing different organelles to investigate Ca²⁺ flux between compartments.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Reagents for Compartment-Specific Calcium Imaging
| Reagent/Category | Specific Examples | Function/Application | Considerations |
|---|---|---|---|
| Chemical Indicators | Fura-2-AM, Fluo-3-AM, Rhod-2-AM | Direct Ca²⁺ detection via fluorescence changes | AM esters require esterase activity; compartmentalization varies by charge |
| GECIs | Cameleon, GCaMP, R-GECO | Genetically-targeted Ca²⁺ sensing | Require transfection/transduction; optimal for long-term studies |
| Targeting Sequences | Mito-roGFP, ER-GCaMP, Nuclear-Localization Signals | Direct GECIs to specific compartments | Verification of localization essential |
| Pharmacological Tools | Ionomycin, Thapsigargin, Cyclopiazonic acid | Manipulate Ca²⁺ levels; empty stores | Specificity varies; use appropriate controls |
| Esterase Promoters | Pluronic F-127 | Enhance AM-ester dye loading | Particularly useful for difficult-to-label cells |
| Microscopy Systems | Confocal, Two-photon, Light-sheet, TIRF | Image acquisition with varying resolution/penetration | Match modality to experimental needs |
Calcium Signaling Pathways and Experimental Workflow
The following diagrams illustrate key calcium signaling pathways and a generalized experimental workflow for compartment-specific calcium imaging.
Diagram 1: Calcium signaling pathways between cellular compartments, showing primary fluxes and regulatory proteins.
Diagram 2: Generalized experimental workflow for compartment-specific calcium imaging studies.
Applications in Disease Research and Drug Development
Compartment-specific calcium imaging provides critical insights into disease mechanisms and therapeutic interventions. In amyotrophic lateral sclerosis (ALS), disturbances in the ER-mitochondria calcium cycle contribute to pathology, with mutant SOD1 and TDP-43 proteins disrupting Ca²⁺ homeostasis and promoting ER stress [70]. In neurodegenerative disorders, dendritic spines compartmentalize calcium to regulate synaptic plasticity, with defective compartmentalization implicated in synaptic dysfunction [74]. For drug development, intestinal organoids enable assessment of nutrient transport, drug uptake, and incretin secretion through Ca²⁺ imaging, complementing traditional in vitro systems [72].
These approaches allow researchers to:
- Screen compounds that modulate compartment-specific Ca²⁺ signaling
- Investigate drug-induced cytotoxicity through mitochondrial Ca²⁺ overload
- Develop therapeutics targeting specific Ca²⁺ transport pathways
- Validate drug mechanisms of action in physiologically relevant model systems
Targeting specific compartments for calcium imaging represents a powerful approach for deciphering the complex spatial organization of Ca²⁺ signaling in live cells. The integration of chemical indicators with genetically-encoded sensors now enables precise monitoring of Ca²⁺ dynamics in the cytosol, ER/SR, and mitochondria with high spatiotemporal resolution. As imaging technologies and analysis algorithms continue to advance, researchers will gain increasingly detailed insights into how compartmentalized Ca²⁺ signals control diverse cellular functions in health and disease. These methodologies provide essential tools for basic research and drug development, particularly for disorders involving disrupted calcium homeostasis such as neurodegenerative diseases, metabolic disorders, and cancer.
Optimizing Detection Fidelity: Tackling Kinetics, Phototoxicity, and Analysis Pitfalls
Calcium ions (Ca²⁺) function as critical intracellular messengers, enabling cells to respond to everything from neural computation to developmental cues with speed and precision [75] [76]. The fidelity with which we can capture these signals depends heavily on the properties of calcium indicators. A fundamental challenge in live-cell imaging is the kinetics-sensitivity trade-off: the widely observed inverse relationship between an indicator's speed (kinetics) and its ability to generate a strong, detectable signal (sensitivity) [6].
This trade-off presents a significant methodological hurdle. While many neuron types exhibit rapid (sub-millisecond) increases in cytoplasmic calcium following action potentials, the fluorescence kinetics of most protein-based sensors are considerably slower, often on the order of hundreds of milliseconds [6]. Consequently, many widely used sensors map relatively static representations of neural information rather than tracking the millisecond-scale dynamics crucial for understanding neural computation [6]. This technical primer examines the biological basis of this trade-off, compares current tools, and outlines strategic approaches for capturing rapid calcium signaling events in live-cell research.
The Biological and Biophysical Basis of the Trade-off
The kinetics-sensitivity trade-off is not merely a technical limitation but is rooted in the molecular biophysics of how calcium indicators function. Genetically Encoded Calcium Indicators (GECIs) like the GCaMP family typically consist of a calcium-binding protein (calmodulin, or CaM), a calmodulin-binding peptide (e.g., RS20), and a fluorescent protein [6]. The binding and unbinding of calcium ions induce a conformational change that modulates fluorescence.
The trade-off often arises from attempts to increase the sensor's affinity for calcium (characterized by a lower dissociation constant, Kd). Higher-affinity sensors bind calcium ions more tightly, which can increase the number of bound ions during a signaling event and thus amplify the fluorescence signal (ΔF/F0). However, this tighter binding also slows the rate at which the sensor releases calcium ions (the dissociation rate, koff), resulting in slower decay kinetics and a reduced ability to track high-frequency spike trains [6] [27].
Furthermore, kinetics are sensitive to mutations at the interface between CaM and its binding peptide, far from the calcium-binding EF hands themselves [6]. This indicates that the post-binding conformational change is a major rate-limiting step. Simply put, engineering efforts that stabilize the bright, calcium-bound state to enhance sensitivity often inadvertently make the transition into and out of this state slower.
Quantitative Comparison of Calcium Indicators
Selecting the right indicator requires a careful balance of kinetics, sensitivity, and brightness. The following tables summarize key performance metrics for a selection of organic dyes and GECIs, providing a reference for informed experimental design.
Table 1: Properties of Common Synthetic Calcium Dyes
| Calcium Indicator | Type | Excitation Max (nm) | Emission Max (nm) | Kd (nM) | Key Applications and Notes |
|---|---|---|---|---|---|
| Fluo-4 [27] | Single wavelength | 490 | 520 | 345 | Fast kinetics; ideal for confocal microscopy; qualitative measurements (F(t)/F(0)) |
| X-rhod-1 [27] | Single wavelength | 580 | 600 | 700 | Red-shifted; reduces phototoxicity & autofluorescence; allows multiplexing |
| Fura-2 [27] | Ratiometric | 340 / 380 | 510 | 145 | Quantitative; corrects for dye concentration & path length; requires UV excitation |
| Indo-1 [27] | Ratiometric | 340 | 405 / 485 | 230 | Ratiometric emission; useful for flow cytometry; can be photo-instable |
Table 2: Properties of Genetically Encoded Calcium Indicators (GECIs)
| Calcium Indicator | Type | Excitation Max (nm) | Emission Max (nm) | Kd (nM) | Reported 1AP ΔF/F0 (%) | Reported Half-Rise Time (ms) |
|---|---|---|---|---|---|---|
| jGCaMP8s [6] | GECI (sensitive) | ~480 | ~510 | ~68 | ~1250 | ~9 |
| jGCaMP8m [6] | GECI (medium) | ~480 | ~510 | ~99 | ~760 | ~5 |
| jGCaMP8f [6] | GECI (fast) | ~480 | ~510 | ~220 | ~330 | ~2 |
| XCaMP-Gf [6] | GECI (fast) | ~480 | ~510 | ~320 | ~90 | ~12 |
Modern Strategies to Overcome the Trade-off
Molecular Engineering of Novel Sensors
Recent protein engineering efforts have successfully broken the traditional kinetics-sensitivity trade-off. The development of the jGCaMP8 series serves as a prime example. Through large-scale screening and structure-guided mutagenesis, researchers targeted the interface between calmodulin and its binding peptide, replacing the traditional RS20 peptide with a peptide from endothelial nitric oxide synthase (ENOSP) [6]. This fundamental redesign yielded sensors with ultra-fast kinetics (half-rise times as low as 2 milliseconds) while maintaining, or even enhancing, sensitivity to single action potentials [6]. The jGCaMP8 family provides tailored variants: jGCaMP8s for maximum sensitivity, jGCaMP8f for maximum speed, and jGCaMP8m as a balanced compromise [6].
Experimental and Computational Workflows
Beyond new reagents, robust experimental and analytical workflows are critical for capturing fast events.
Diagram 1: A generalized workflow for capturing rapid calcium signaling events, highlighting key decision points to manage the kinetics-sensitivity trade-off.
Multiplexed Imaging and Analysis
Another powerful strategy involves multiplexing, or simultaneously imaging multiple biological parameters. This can help infer causal relationships within signaling networks. Key approaches include:
- Spectral Multiplexing: Using biosensors with spectrally distinct fluorophores, such as combining green/yellow and red fluorescent indicators, to simultaneously track calcium and another signaling molecule (e.g., cAMP) [77].
- Temporal and Spatial Multiplexing: Leveraging photochromic proteins that switch states or localizing biosensors to specific subcellular compartments to distinguish signals [77].
- Computational Deconvolution: The improved linearity of newer sensors like jGCaMP8 allows for more robust deconvolution of fluorescence transients into underlying action potentials, effectively using algorithms to infer faster events from the sensor's kinetic response [6].
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Research Reagent Solutions for Calcium Imaging
| Reagent / Material | Function / Description | Example Use Case |
|---|---|---|
| jGCaMP8 Series [6] | A 3rd-generation GECI family offering variants optimized for speed (8f), sensitivity (8s), or a balance (8m). | In vivo imaging of neural populations to track individual spikes at high frequencies (up to 50 Hz). |
| XCaMP Series [6] | A family of GECIs utilizing a camkkap peptide instead of RS20/ENOSP, providing an alternative design for fast kinetics. | Fast calcium imaging where green and red indicators are needed for multiplexing. |
| Fluo-4 AM [27] | A cell-permeant, single-wavelength synthetic dye. Easy loading into cells; bright fluorescence upon calcium binding. | High-throughput screening of cellular activity in plate readers or confocal microscopy. |
| Fura-2 AM [27] | A cell-permeant, ratiometric synthetic dye. Its signal is self-referencing, correcting for variations in dye concentration. | Quantitative measurement of absolute calcium concentrations in single cells. |
| CoolLED pE-340fura [27] | An LED light source providing specific wavelengths (340 nm & 380 nm) required for ratiometric dyes like Fura-2. | Stable, high-speed excitation for quantitative ratiometric calcium imaging. |
The kinetics-sensitivity trade-off has long been a defining challenge in the field of calcium imaging. However, as this guide illustrates, the field is moving beyond this limitation through innovative molecular engineering, sophisticated experimental designs, and powerful computational analyses. The development of sensors like the jGCaMP8 series, which provide both ultra-fast kinetics and high sensitivity, is a testament to this progress. For the researcher, the path forward involves a strategic choice: selecting the indicator whose specific blend of kinetic and sensitivity parameters best matches the biological question at hand, while leveraging complementary methodological workflows to extract the maximum amount of information from the resulting signals. As protein engineering continues to refine biosensors and computational tools become ever more integral to experimental biology, our capacity to decode the rapid, calcium-based language of cells will only continue to grow.
Calcium ions (Ca²⁺) function as ubiquitous intracellular messengers, regulating processes from neurotransmission and muscle contraction to gene expression and cell death [35] [78]. The study of Ca²⁺ signaling in live cells relies on advanced imaging techniques, primarily using fluorescent indicators. However, the very tools that enable these observations—chemical dyes, genetically encoded indicators, and the microscopy systems themselves—can introduce significant experimental artifacts by perturbing the native cellular environment. Three primary sources of perturbation are the buffering of native Ca²⁺ signals by indicators, phototoxicity from light exposure during imaging, and the ectopic expression of sensor proteins. These artifacts can distort the physiological signals researchers aim to measure, leading to erroneous conclusions. This guide provides an in-depth technical framework for researchers and drug development professionals to identify, quantify, and minimize these perturbations within the context of Ca²⁺ signaling research, ensuring that observed dynamics faithfully represent underlying biology.
Calcium Buffering by Synthetic and Genetically Encoded Indicators
A fundamental challenge in Ca²⁺ imaging is that the introduction of a Ca²⁺-binding indicator adds buffering capacity to the cytoplasm, which can alter the kinetics and amplitude of the native Ca²⁺ transients.
Mechanisms of Artifact Generation
Genetically Encoded Calcium Indicators (GECIs), such as GCaMPs, operate by coupling a Ca²⁺-sensing calmodulin (CaM) domain to a fluorescent protein. The CaM domain binds to Ca²⁺, inducing a conformational change that alters fluorescence. However, this introduced CaM, along with its binding peptide (e.g., M13, ckkap), competes with endogenous CaM targets for available Ca²⁺ and can interfere with native Ca²⁺-dependent signaling pathways [35]. This is particularly problematic in signaling microdomains where precise, localized Ca²⁺ transients are critical for function. The high-affinity binding of these sensors means they not only report but also sequester Ca²⁺, smoothing out rapid fluctuations and reducing the peak amplitude of signals.
Strategies for Minimizing Buffering Artifacts
- Use the Lowest Practical Indicator Concentration: The buffering effect is directly proportional to the concentration of the indicator. Titrate the expression level or loading concentration to the minimum required for an acceptable signal-to-noise ratio.
- Select Low-Affinity Indicators for High Ca²⁺ Environments: In compartments with high resting Ca²⁺, such as the endoplasmic reticulum (ER, ∼2000 times higher than cytosol) or mitochondria, standard high-affinity indicators are saturated and unresponsive. Specialized low-affinity sensors have been engineered for these environments. For example, ER-HaloCaMP is a chemigenetic indicator with an affinity (EC₅₀) tuned to ∼115 µM, making it suitable for reporting dynamics within the ER lumen without being saturated at resting concentrations [79].
- Employ Chemigenetic Sensors for Tunability: Chemigenetic sensors, like the HaloCaMP toolkit, offer a flexible solution. These sensors consist of a genetically encoded protein tag (e.g., HaloTag) that binds synthetically to a cell-permeable fluorescent dye-ligand. This allows the color and photophysical properties of the sensor to be tuned by using different dye-ligands without the need to re-engineer the protein, providing a versatile approach to optimize imaging parameters and minimize perturbation [79].
Table 1: Strategies to Mitigate Calcium Buffering Artifacts
| Strategy | Mechanism | Example Tools | Application Context |
|---|---|---|---|
| Minimal Expression/Loading | Reduces total Ca²⁺-binding sites | Titrating GCaMP DNA transfection amount; using low dye concentrations | All live-cell imaging experiments |
| Low-Affinity Sensors | Prevents saturation in high [Ca²⁺] environments | ER-HaloCaMP (EC₅₀ ~115 µM), Mito-HaloCaMP | ER, mitochondrial matrix, synaptic clefts |
| Chemigenetic Indicators | Decouples sensor affinity from color; allows optical optimization | HaloCaMP series with JF dye-ligands | Multiplexed imaging; deep-tissue imaging |
Phototoxicity: Mechanisms, Consequences, and Mitigation
Phototoxicity refers to light-induced damage that disrupts cellular physiology, creating artifacts that can confound experimental results. It is a major, often overlooked, source of perturbation in live-cell microscopy.
The Molecular Cascade of Photodamage
The primary mechanism of phototoxicity involves the generation of reactive oxygen species (ROS). When excitation light illuminates fluorescent molecules (indicators or endogenous chromophores), they can transfer energy to molecular oxygen, creating singlet oxygen (¹O₂) and other ROS [80]. These highly reactive molecules cause widespread damage, including lipid peroxidation, protein oxidation, and DNA strand breaks [81] [80]. A 2024 study systematically illuminated intestinal organoids (enteroids) and used RNA-seq to reveal that even low-dose light illumination (71 J/cm²) induced significant abnormalities in gene expression related to ROS response, metabolism, mitosis, and immune pathways before any morphological changes were visible. High-dose illumination (1063 J/cm²) led to overt apoptosis and functional failures in structure-forming ability and Paneth cell secretion [81].
Quantitative Light Dosage and Cellular Impact
Understanding and monitoring light dosage is critical for experimental design. The table below summarizes key light dose thresholds and their observed effects, highlighting the sensitivity of cellular systems.
Table 2: Quantitative Effects of Light Exposure in Live-Cell Imaging
| Light Dose (J/cm²) | Wavelength | Observed Cellular Consequences | Experimental Model |
|---|---|---|---|
| 71 J/cm² (Low-dose) | 488/647 nm | mRNA expression changes for ROS, metabolism, mitosis; no visible morphology change | Mouse enteroids [81] |
| 1063 J/cm² (High-dose) | 488/647 nm | Activation of apoptosis-related genes; failure of structure formation & granule secretion | Mouse enteroids [81] |
| ~10 J/cm² (Limit for stained cells) | e.g., 488 nm | Common limit for non-phototoxic conditions with dyes/GFP; allows ~100 wide-field images | General guideline [80] |
A Practical Workflow for Mitigating Phototoxicity
The following diagram outlines a systematic workflow to minimize phototoxicity, from experimental setup to image acquisition and validation.
Advanced Methodologies: Light-Sheet Fluorescence Microscopy (LSFM)
For long-term, high-resolution 3D imaging, Light-Sheet Fluorescence Microscopy (LSFM) is a superior approach. Unlike confocal microscopy, which illuminates the entire sample to image a single plane, LSFM illuminates only the focal plane with a thin "sheet" of light. This confines photobleaching and photodamage to the plane being imaged, drastically reducing the total light dose delivered to the sample. This allows for a much higher number of images to be acquired within the "light dose budget," making it ideal for imaging rapid Ca²⁺ dynamics in sensitive samples like embryos or organoids over extended periods [80].
Ectopic Expression Artifacts and Genetic Perturbation
The introduction and overexpression of genetically encoded sensors can disrupt cellular physiology through several mechanisms unrelated to their Ca²⁺-sensing function.
Case Study: The brush Mutant and Quantitative Competition
A seminal study of the brush mutant in Lotus japonicus provides a powerful example of ectopic expression interference. The brush phenotype, characterized by stunted roots and aborted rhizobial infection, was caused by a point mutation in a cyclic nucleotide-gated channel (CNGC) subunit, creating a "leaky" channel [82]. Surprisingly, introducing a wild-type genomic copy of the gene did not rescue the phenotype. The researchers discovered that the mutant and wild-type channel subunits compete for assembly into tetrameric channels. The mutation exhibited a quantitative, dosage-dependent effect; only overexpression of the wild-type gene or related redundant CNGC subunits could suppress the mutant phenotype. This case highlights how a mutated protein can exert a dominant-negative effect through stoichiometric competition, a phenomenon that can be mirrored by the overexpression of any exogenous protein, including GECIs [82].
Overexpression and Mislocalization Artifacts
High-level overexpression of GECIs can overwhelm cellular machinery, leading to protein aggregation, endoplasmic reticulum stress, and disruption of organelle function. Furthermore, adding targeting sequences (e.g., for mitochondria, ER, or plasma membrane) does not guarantee perfect localization. Mislocalized sensors can report Ca²⁺ from incorrect compartments, leading to misinterpretation of data. For instance, a sensor intended for the mitochondrial matrix that partially localizes to the cytoplasm will report a conflated signal, obscuring true compartment-specific dynamics.
Strategies for Minimizing Genetic Perturbation
- Use Endogenous Promoters or Knock-In Strategies: Drive expression of the GECI using its native promoter or, ideally, knock the GECI sequence into the native locus of a "housekeeping" gene to achieve physiological expression levels. This avoids the gross non-physiological overexpression common with viral promoters.
- Validate with Loss-of-Function Controls: As demonstrated by the brush study, a critical control is to show that a null mutant of the sensor gene does not recapitulate the phenotype observed in sensor-expressing cells [82]. This confirms that the phenotype is due to the sensor's function and not its physical presence or expression.
- Characterize Expression and Localization Rigorously: Use immunostaining or fused tags to confirm the sensor localizes correctly and is expressed at reasonable levels without forming aggregates.
- Employ Soma-Targeted Sensors for Neuronal Imaging: In neuroscience, soma-targeted GECIs like SomaFRCaMPi reduce neuropil contamination—signal arising from surrounding axons and dendrites that can obscure soma-specific fluorescence. This improves single-cell resolution and can reduce the sensor load in sensitive neuronal processes [83].
The Scientist's Toolkit: Key Reagents and Methodologies
Table 3: Essential Research Reagents and Tools for Minimizing Perturbation
| Tool / Reagent | Function | Key Feature / Application |
|---|---|---|
| FR-GECO1a/c [33] | Far-red genetically encoded Ca²⁺ indicator | Excitation/emission (~596/646 nm) within optical window; reduced scattering & phototoxicity. |
| ER-/Mito-HaloCaMP [79] | Chemigenetic organellar Ca²⁺ indicator | Tunable color with JF dyes; low affinity optimized for ER/mitochondria [Ca²⁺]. |
| SomaFRCaMPi [83] | Soma-localized red GECI | Reduces neuropil contamination in neuronal population imaging. |
| Light-Sheet Microscopy (LSFM) [80] | Imaging technique | Illuminates only the focal plane, drastically reducing total light dose & phototoxicity. |
| RNA-Sequencing (RNA-seq) [81] | Transcriptome analysis | Validates subtle phototoxic effects by revealing changes in stress pathway gene expression. |
| Low-Affinity GECI Mutants | Engineered sensors | Mutations in Ca²⁺-coordinating sites lower affinity for use in high [Ca²⁺] compartments. |
Faithful recording of calcium signals in live cells requires a vigilant and multifaceted approach to experimental design. Researchers must treat their measurement tools not as passive observers, but as active participants in the cellular environment that can distort the very phenomena they are designed to study. By understanding the mechanisms of buffering, phototoxicity, and ectopic expression, and by implementing the strategies outlined in this guide—using minimal sensor expression, leveraging red-shifted and chemigenetic tools, adopting light-efficient microscopy, and employing rigorous genetic and functional controls—scientists can minimize cellular perturbation. This disciplined approach ensures that the observed calcium dynamics are a true reflection of cellular physiology, thereby strengthening the foundation of discovery in basic research and drug development.
In the study of cellular processes, calcium signaling is a fundamental mechanism underpinning everything from neurotransmission to gene expression. The ability to detect these signals in live cells via calcium imaging has revolutionized our understanding of cellular communication. However, the raw data captured from these experiments is fraught with noise and movement artifacts. A robust data analysis pipeline is, therefore, not merely a supplementary step but a critical component for extracting biologically meaningful information. This technical guide delves into the core stages of this pipeline—motion correction, region of interest (ROI) extraction, and background subtraction—framed within the context of modern calcium signaling detection methods.
Motion Correction: Stabilizing the Dynamic Cell
The first challenge in analyzing calcium imaging data is compensating for sample movement. In live-cell imaging, particularly in vivo, motion occurs from physiological processes like heartbeat, respiration, or the animal's own movement. Failure to correct for this motion misaligns cellular signals across time, leading to inaccurate measurements.
Core Methodologies and Algorithms
Motion correction aims to apply a spatial transformation to each frame of the video sequence to align it with a common reference. The two primary approaches are rigid registration and non-rigid alignment.
Rigid Registration: This method corrects for global movement in the x-y plane, including translation and rotation. It is computationally efficient and is often sufficient for well-stabilized samples. Tools like the Multicellular analysis Calcium Imaging (MCA) toolbox for ImageJ implement rigid registration to correct for motion artifacts [84]. The process typically involves:
- Generating a reference image, often the mean projection or a specific frame.
- For each frame, calculating the cross-correlation with the reference image.
- Determining the optimal translational (and sometimes rotational) shift that maximizes this correlation.
- Applying the shift to realign the frame.
Non-Rigid Alignment: Chronic imaging over days or weeks, common in studies of learning and memory, often involves non-rigid brain deformations. These require more sophisticated correction methods. The CaliAli suite addresses this by using a diffeomorphic Demons algorithm to calculate smooth and continuous displacement fields that warp one image to another, correcting for complex distortions across sessions [85]. This method is particularly powerful for aligning data from freely moving animals across multiple sessions, ensuring the same neuron can be tracked over long periods.
Advanced Strategies: Leveraging Stable Landmarks
A significant innovation in motion correction, especially for one-photon microscopy, is the use of intrinsic biological landmarks. CaliAli, for instance, repurposes Hessian filters from ocular imaging to selectively enhance the contrast of dark, elongated blood vessels in the field of view [85]. These vessels provide a stable, high-contrast structural framework that is more reliable for alignment than neuronal signals alone, which can change from session to session. The software employs a multiscale approach, first aligning blood vessels at a coarse level to handle large deformations before refining the alignment using neuronal projections at higher resolutions [85].
Table 1: Key Motion Correction Algorithms and Their Applications
| Algorithm/Approach | Type of Correction | Best Suited For | Example Toolboxes |
|---|---|---|---|
| Rigid Registration | Global (Translation, Rotation) | Head-fixed imaging; stable cell cultures | MCA [84], Minian [86] |
| Non-Rigid Diffeomorphic | Local, Elastic Deformations | Chronic imaging; freely moving animals with long-term experiments | CaliAli [85] |
| Landmark-Based (Vessels) | Non-Rigid | One-photon miniscope data with visible vasculature | CaliAli [85] |
The following diagram illustrates the strategic multi-level alignment workflow that leverages both blood vessels and neuronal projections for superior motion correction.
Once the video data is stabilized, the next step is to identify the specific cellular compartments generating the calcium signals. This process, known as ROI extraction, segments the video data into spatially distinct components corresponding to individual neurons or subcellular structures.
Segmentation Strategies for Diverse Structures
The choice of ROI extraction method depends heavily on the imaging target—whether it is neuronal somata, dendrites, or axons.
Soma Segmentation with CNMF: The Constrained Non-negative Matrix Factorization (CNMF) approach is a powerful and widely used method. It models the entire video data as a product of two matrices: a spatial matrix containing the footprints (shapes and locations) of each neuron, and a temporal matrix containing the calcium fluorescence trace of each neuron over time [86]. This method is effective at demixing signals from overlapping or nearby neurons, a common issue in dense neural tissue. Minian and CaImAn are popular toolboxes that implement CNMF and its variants [86].
Subcellular Segmentation with Spectral and Frequency Analysis: Extracting signals from thin axons and dendrites is particularly challenging due to their low signal-to-noise ratio (SNR). The SUBPREP pipeline addresses this by applying a Fast Fourier Transform (FFT) on smoothed calcium traces. It then uses bandpass filtering (e.g., 0.05–0.12 Hz) to select ROIs whose frequency profiles match the known power band of genuine calcium transients, effectively separating real signals from noise [87]. Similarly, the SpecSeg toolbox identifies ROIs by analyzing the temporal cross-correlations of low-frequency components between each pixel and its neighbors, a method that works intuitively for both neuronal cell bodies and neurites [88].
Leveraging Machine Learning: Traditional algorithms can struggle with varied cell morphologies. The MCA toolbox integrates a custom model trained in Cellpose 2.0, a generalist algorithm for cellular segmentation, specifically tuned for segmenting nuclei expressing pan-neuronal nuclear-localized GCaMP in zebrafish [84]. This demonstrates a move towards adaptable, trained models for accurate ROI detection.
The Challenge of ROI Grouping
In subcellular imaging, a single axon or dendrite may be represented by multiple disconnected ROIs as it traverses the field of view. SUBPREP addresses this by employing hierarchical or k-means clustering on the temporal activity profiles of ROIs [87]. ROIs with highly correlated calcium transients are grouped together, indicating they likely belong to the same neural process, which improves SNR and prevents overrepresentation of single neurons in the data.
Table 2: ROI Extraction Techniques for Different Biological Targets
| Technique | Underlying Principle | Optimal Use Case | Strengths |
|---|---|---|---|
| CNMF(-E) | Matrix Factorization | Densely packed neuronal somata (2P & 1P) | Demixes overlapping signals; models background |
| Spectral (SpecSeg) | Low-frequency Cross-correlation | Neurons and neurites in chronic data | Intuitive; works for both 1P and 2P data [88] |
| Frequency (SUBPREP) | FFT & Bandpass Filtering | Low-SNR axons and dendrites | Effectively isolates physiological signals from noise [87] |
| Machine Learning (Cellpose) | Pretrained Neural Network | Consistent cellular structures (e.g., nuclei) | High accuracy with a well-trained model [84] |
The workflow for ROI extraction and signal processing, from initial segmentation to final trace generation, is summarized below.
Background Subtraction: Isolating the True Signal
The fluorescence measured from an ROI contains not only the desired signal from the neuron of interest but also a contaminating "background" signal. This background originates from out-of-focus fluorescence from nearby cells and neuropil, as well as extracellular autofluorescence. Accurate background estimation and subtraction is crucial for obtaining a true representation of cellular calcium dynamics.
Modeling the Background Component
Structured Background with CNMF-E: In one-photon miniscope data, a significant source of background is out-of-focus fluorescence. The CNMF-E algorithm extends CNMF by explicitly modeling this background as a spatially structured component. It uses a ring model that captures the diffuse, surrounding glow typical of single-photon imaging, allowing for its effective separation from the focal neuronal signals [86].
Morphological Operations for Background Estimation: The MIN1PIPE pipeline, another tool for miniscope data, uses morphological operations during preprocessing to identify and remove the background fluorescence [86]. This approach involves creating a background estimate by applying operations that capture the low-frequency, slowly varying components of the image, which are then subtracted from the original data.
Neuropil Subtraction: A common and straightforward method for background subtraction, especially in two-photon data, is neuropil subtraction. This involves defining a annulus around each ROI that excludes other identified cells. The fluorescence signal from this ring is considered the local background, which is then scaled by a factor (e.g., 0.7) and subtracted from the signal from the central ROI. This method corrects for contaminating signals from the dense network of processes surrounding the cell body.
The Scientist's Toolkit: Essential Research Reagents and Tools
The following table catalogues key reagents, software tools, and analytical packages that form the foundation of a modern calcium imaging data analysis pipeline.
Table 3: Research Reagent Solutions for Calcium Imaging Analysis
| Item Name | Type | Primary Function | Technical Notes |
|---|---|---|---|
| GCaMP6s/8 | Genetically Encoded Calcium Indicator (GECI) | Converts neuronal spiking activity into fluorescent signals | High sensitivity; widely used; available in axon-targeted versions (e.g., AAV9-axon-GCaMP6s) [87] |
| FR-GECO1 | Far-red GECI | Enables deep-tissue imaging and multicolor experiments with other green probes | Excitation/Emission max at ~596/~644 nm; resides in the optical window for better tissue penetration [33] |
| CaliAli | Software Suite | Corrects inter-session misalignments in chronic 1P imaging | Uses blood vessels and diffeomorphic demons for robust non-rigid registration [85] |
| Minian | Software Pipeline | Extracts calcium signals from 1P miniscope data | Open-source; low memory demand; interactive visualization for parameter tuning [86] |
| Suite2P | Software Pipeline | Performs initial ROI identification and basic processing | Foundation for many pipelines; can be extended with custom post-processing [87] |
| CalciumNetExploreR (CNER) | R Package | Performs network analysis on extracted calcium traces | Analyzes functional connectivity and topology; requires pre-extracted traces [14] |
The path from a raw, flickering calcium imaging video to a precise quantitative understanding of cellular signaling is paved by a rigorous and multifaceted data analysis pipeline. Motion correction stabilizes the dynamic world of the live cell, ROI extraction pinpoints the origins of its signals, and background subtraction purifies these signals from contamination. As calcium imaging continues to evolve, pushing into deeper tissues and longer timescales, the tools and methods described here—from the demon-based non-rigid alignment in CaliAli to the spectral segmentation of SpecSeg—will remain indispensable. Mastering this pipeline is not a mere technicality; it is a fundamental requirement for any researcher aiming to faithfully decode the intricate language of calcium in living systems.
Calcium ions (Ca²⁺) function as ubiquitous intracellular messengers, regulating processes from neurotransmission and muscle contraction to gene expression and cell proliferation [35] [18]. The detection of transient increases in intracellular calcium—rapid shifts in concentration that form the basis of this signaling system—is fundamental to understanding cellular communication. Defining activity thresholds constitutes a central challenge in this process, as the accuracy of threshold determination directly impacts the reliability, reproducibility, and biological validity of calcium imaging data [35]. The inherent variability of calcium transients, especially their low signal-to-noise ratio (SNR) in key experimental contexts like synaptic events or embryonic development, makes simplistic thresholding approaches insufficient [35] [89]. This technical guide examines the core challenges in transient detection and outlines robust, modern methodologies for establishing accurate activity thresholds, providing a critical resource for researchers and drug development professionals working within the broader field of calcium signaling detection.
Core Challenges in Calcium Transient Detection
The first challenge arises from the biological and physical nature of the signals themselves. Calcium transients are not uniform; they exhibit a vast diversity in form. Spikes are high-amplitude, short-duration events restricted to single cells, while waves are lower-amplitude, longer-duration events that propagate through multiple cells [35]. This inherent variability is compounded by technical factors. In developing systems, such as embryonic neural tissue, calcium activity is far more sporadic and irregular than in mature cells, defying simple, consistent analysis [35]. Furthermore, the choice of calcium indicator—each with its own affinity, brightness, and kinetics—profoundly affects the measured fluorescence. For instance, the GCaMP6f indicator offers high affinity and fast kinetics, but signals from miniature Synaptic Calcium Transients (mSCTs) monitored with such sensors still exhibit low SNR and wide morphological variability [90] [89].
Methodological Pitfalls in Threshold Determination
A primary analytical challenge is the lack of standardization in how baselines and thresholds are defined across studies. The method used for signal preprocessing and baseline definition can dramatically alter the resulting data interpretation [35]. Applying a single, global threshold to an entire dataset often yields inconsistent results, as baseline activity and noise characteristics can vary between cells and even within different regions of the same cell [91] [89]. Threshold-based methods struggle particularly with small, propagating events that change size and shape over time, leading to either a high rate of false positives (detecting noise as events) or false negatives (missing genuine biological events) [90] [89]. These pitfalls underscore the necessity for more sophisticated, adaptive, and validated detection strategies.
Established and Emerging Methodologies for Threshold Detection
Traditional Statistical and Parameter-Based Methods
Traditional methods often rely on statistical properties of the fluorescence signal to set a detection threshold. A common approach involves an iterative algorithm that performs Gaussian fitting to the histogram of all data points in a trace. The data typically forms a skewed distribution with a large peak at the baseline and a long tail representing transient events. The baseline mean and standard deviation (SD) are derived from this fit, and a threshold is set at a defined number of SDs above the baseline mean (e.g., 4 SD), with the start and end times of an event defined at a lower value (e.g., 2 SD) [92]. Another robust method involves calculating a threshold for each frame as the median plus three times the interquartile range of the ΔF/F₀ values, which is less sensitive to outliers than the mean/SD approach [90]. These methods often require subsequent filtering steps, such as excluding regions of interest (ROIs) that consist of only a single pixel (likely camera noise) or that occur outside a defined cellular mask [90].
Machine Learning and Deep Learning-Driven Approaches
Emerging paradigms leverage machine learning to overcome the limitations of fixed-threshold models. Supervised deep learning (DL) models, such as 3D U-Net and StarDist-3D, learn to detect and segment calcium transients directly from spatio-temporal (x, y, t) imaging data without requiring user-defined thresholds or a priori assumptions about event morphology [89]. These data-driven approaches excel at identifying small, variable events like mSCTs. A significant innovation is Positive-Unlabeled (PU) learning, which trains DL models using a set of confirmed positive events (mSCTs) and a large set of unlabeled data. This is particularly powerful in calcium imaging, where genuine transients are scarce and annotating all negative instances is impractical. PU learning significantly enhances model performance by leveraging the abundance of unlabeled data to better distinguish signals from background fluctuations [89]. For larger-scale neuronal activity analysis, tools like CalTrig integrate machine learning models including Gated Recurrent Units (GRU), Long Short-Term Memory (LSTM), and Transformers. Among these, the GRU model has demonstrated the highest predictability and computational efficiency for detecting Ca²⁺ transients from miniScope data in freely moving animals [91].
Table 1: Comparison of Calcium Transient Detection Methods
| Method Category | Key Example | Underlying Principle | Key Advantages | Key Limitations |
|---|---|---|---|---|
| Statistical Thresholding | Standard Deviation-based [92] | Iterative Gaussian fitting of fluorescence histogram to define baseline & threshold. | Computationally simple, intuitive, requires no training data. | Sensitive to noise, struggles with low-SNR and propagating events. |
| Statistical Thresholding | Median + IQR [90] | Uses median and interquartile range of ΔF/F₀ to set a per-frame threshold. | Robust to outliers in the signal. | Still requires post-hoc filtering of false positives. |
| Machine Learning | CalTrig (GRU/LSTM) [91] | Recurrent Neural Networks model temporal dynamics of calcium traces. | High computational efficiency; stable performance across subjects/brain regions. | Requires a training dataset; model performance depends on training data quality. |
| Deep Learning | 3D U-Net with PU Learning [89] | 3D convolutional network trained with positive and unlabeled data for segmentation. | Data-driven; superior for small, variable events (mSCTs); no need for explicit thresholds. | High computational demand; requires significant expertise to implement and train. |
Integrated GUI Tools for Flexible Analysis
To make advanced detection methods accessible to non-programmers, graphical user interface (GUI) tools have been developed. CalTrig is an open-source tool that integrates multiple data streams (Ca²⁺ imaging videos, traces, cell footprints, behavior) and, crucially, provides a single platform for applying manual, parameter-based, and machine learning-based detection methods. This allows researchers to compare methods and choose the most appropriate one for their specific application, thereby bridging the gap between raw data extraction and final biological insight [91]. Similarly, SICT is a MATLAB-based program that automates the detection of fast, low-amplitude transients and provides a GUI for supervised inspection, reducing manual labor to less than 10% while maintaining quality control [90].
Experimental Protocols for Threshold Determination
Protocol: Standard Statistical Threshold Detection for Neuronal Somas
This protocol is adapted from methods used to detect somatic calcium transients and correlate them with local field potentials [92].
- Signal Preprocessing: Begin by correcting the raw ΔF/F trace for slow trends, such as photobleaching or focal drift, using a smoothing algorithm. A robust locally-weighted regression (e.g., with a window spanning 25% of the file duration) is effective for this step. Subtract this smoothed baseline from the original trace to obtain a baseline-corrected ΔF/F signal.
- Baseline Estimation: Create a histogram of all data points from the baseline-corrected trace. For most cells, this distribution is skewed. Perform an iterative Gaussian fitting:
- First Iteration: Apply a double-Gaussian fit to the entire histogram.
- Second Iteration: Use the higher amplitude Gaussian's SD from the first fit to constrain a single-Gaussian refinement fit focused only on the baseline peak. The mean of this final Gaussian is your baseline mean (μ), and its standard deviation is the baseline SD (σ).
- Threshold Setting and Event Detection: Set the detection threshold at μ + 4σ. To define the start and end times of each transient, use a lower threshold of μ + 2σ. The event begins when the signal crosses above μ + 2σ and ends when it decays back below μ + 2σ.
- Validation: For critical applications, validate a subset of detected events by manual inspection to ensure the threshold parameters are appropriately tuned for the specific experimental conditions.
Protocol: Automated Detection with Supervised Inspection (SICT)
This protocol is designed for detecting smaller, spontaneous calcium transients (SCTs) in cultured neurons [90].
- Data Preparation and Filtering: Acquire time-lapse movies (e.g., at ~29 Hz) of cells expressing a fast indicator like GCaMP6f. Apply a 3D (x, y, t) Gaussian filter to the data to reduce noise.
- Calculate ΔF/F₀: Compute ΔF/F₀ using a moving average for F₀ to optimize for the detection of fast rises (this is unsuitable for slow fluctuations).
- Apply Adaptive Threshold: For each frame, calculate the threshold as the median plus 3 times the interquartile range of all pixel values in that frame.
- Identify Regions of Interest (ROIs): Group all super-threshold pixels that are adjacent in space or time into 3D ROIs, each representing a putative calcium event.
- Filter ROIs:
- Exclude ROIs consisting of a single pixel (likely camera hot pixels).
- (Optional) Exclude ROIs that fall outside a defined cellular mask (e.g., areas with above-average intensity) to focus on biological structures.
- Supervised Inspection: Use the built-in GUI to visually inspect each automatically detected ROI. Manually classify each event as a true positive or false positive. This step combines computational efficiency with expert validation.
The following workflow diagram illustrates the key decision points in a robust transient detection pipeline, integrating both traditional and machine-learning approaches.
Calcium Transient Detection Workflow
The Scientist's Toolkit: Essential Reagents and Tools
Table 2: Research Reagent Solutions for Calcium Imaging
| Tool Category | Specific Example | Key Function & Characteristics | Typical Application Context |
|---|---|---|---|
| Ratiometric Dyes | Fura-2 AM [22] [39] | Cell-permeant dye; excitation shifts from 340 nm (Ca²⁺-bound) to 380 nm (unbound). Allows quantitative [Ca²⁺] calculation, minimizes artifacts. | Live-cell imaging of various cell types; GPCR and calcium channel studies. |
| Intensity-Based Dyes | Fluo-4 AM [22] | Cell-permeant dye; >100-fold fluorescence increase upon Ca²⁺ binding; no resting signal. Very bright and sensitive. | High-throughput screening, flow cytometry, detecting large [Ca²⁺] changes. |
| Genetically Encoded Indicators | GCaMP6f/8f [91] [89] | Genetically encoded Ca²⁺ indicator (GECI); green fluorescence increases with Ca²⁺. Targetable to specific cell types/subcellular compartments. | In vivo imaging in transgenic animals; long-term studies in cultured neurons/organoids. |
| Analysis Software | CalTrig [91] | Open-source GUI tool; integrates ML models (GRU/LSTM) and parameter-based detection for miniScope data. | Analysis of in vivo Ca²⁺ imaging from freely moving animals. |
| Analysis Software | SICT [90] | MATLAB-based GUI; automates detection of low-amplitude SCTs and allows supervised inspection. | Detection of spontaneous synaptic transients in cultured neurons. |
Defining accurate activity thresholds for calcium transient detection remains a nuanced but surmountable challenge. While traditional statistical methods provide a foundational approach, their limitations in handling low-SNR and complex biological signals are increasingly evident. The field is moving toward a new paradigm dominated by machine learning and deep learning techniques, which offer data-driven, adaptive, and highly accurate detection without relying on rigid, user-defined thresholds. The integration of these advanced algorithms into user-friendly GUI tools like CalTrig and SICT, coupled with robust experimental protocols, empowers researchers to extract deeper, more reliable biological insights from their calcium imaging data. This progression is essential for advancing our understanding of the "calcium code" in both health and disease, ultimately accelerating discovery in basic research and drug development.
Calcium ions (Ca²⁺) function as a ubiquitous intracellular messenger, regulating processes from neurotransmission and muscle contraction to gene expression and cell development [46] [93]. The visualization of these dynamics via calcium imaging provides a powerful window into cellular activity. The most common metric in calcium imaging is the normalized fluorescence change, ΔF/F0, which reports relative changes in intracellular calcium. While ΔF/F0 is invaluable for detecting the timing and pattern of calcium signals, it is an arbitrary unit that does not reveal the actual concentration of calcium ions [94].
Moving from this relative metric to absolute concentration values is a critical step for a deeper, more mechanistic understanding. Quantification allows for direct comparison between different cell types, experimental conditions, and laboratories. It is essential for precisely modeling calcium-dependent processes, understanding the activation thresholds of calcium-sensitive enzymes, and accurately assessing pharmacological effects in drug development [93] [95]. This guide details the principles and protocols for performing this vital calibration, enabling researchers to translate qualitative observations into quantitative biological insights.
Theoretical Foundations: From Fluorescence to Concentration
The Meaning of ΔF/F0
The calculation of ΔF/F0 begins with extracting a fluorescence trace, F(t), from a region of interest (ROI) within a cell. The baseline fluorescence, F0, represents the fluorescence intensity when the cell is at its resting calcium concentration. ΔF/F0 is then calculated as (F(t) - F0)/F0 [94].
This normalization serves two key purposes: First, it accounts for variability in baseline brightness between cells caused by differences in dye loading or expression levels of genetically encoded indicators. Second, for traditional single-binding-site chemical dyes, ΔF/F0 was historically designed to be proportional to the change in the number of calcium-bound indicator molecules, providing a link to the underlying calcium concentration [94].
The Relationship Between Fluorescence and [Ca²⁺]
The fluorescence of most calcium indicators changes upon binding Ca²⁺ ions. The relationship between the concentration of free calcium ([Ca²⁺]) and the fluorescence signal is described by a binding curve.
For Single-Wavelength Indicators (e.g., Fluo-4, OGB-1, GCaMP): The fluorescence intensity (F) increases as [Ca²⁺] increases. The relationship follows a saturating curve, which can be described for indicators with a single binding site as:
Where
F_minis the fluorescence in calcium-free conditions,F_maxis the fluorescence at saturating calcium, andK_dis the dissociation constant [95].For Ratiometric Indicators (e.g., Fura-2, Indo-1): These indicators exhibit a spectral shift upon Ca²⁺ binding. Fura-2, for instance, is excited at 340 nm when bound to Ca²⁺ and at 380 nm when unbound. The ratio of fluorescence (R = F₃₄₀/F₃₈₀) is used, which is largely independent of dye concentration and cell thickness. The relationship is given by:
Where
R_minandR_maxare the ratios at zero and saturating [Ca²⁺], andβis the ratio of fluorescence intensities at 380 nm in zero and saturating [Ca²⁺] [39] [96].For Genetically Encoded Indicators (e.g., GCaMP): GCaMP and its variants are based on a single fluorescent protein and calmodulin, which has multiple cooperative binding sites. Their behavior is better described by the Hill equation:
Where
nis the Hill coefficient representing cooperativity (e.g., n≈2.3 for GCaMP6f) [94]. This results in a sigmoidal relationship between ΔF/F0 and [Ca²⁺], which provides high sensitivity over a specific concentration range but introduces nonlinearity.
Table 1: Key Parameters for Common Genetically Encoded Calcium Indicators (GECIs)
| Indicator | Approximate K_d (nM) | Hill Coefficient (n) | Linearity Considerations |
|---|---|---|---|
| GCaMP6f | ~375 | ~2.3 | Sigmoidal curve; nonlinear especially at high [Ca²⁺] |
| GCaMP7f | ~150 | ~3.1 | Highly sigmoidal; sensitive to single action potentials |
| GCaMP8s | ~46 | ~2.2 | High affinity, saturates quickly with multiple spikes |
| jGCaMP7s | ~68 | ~2.8 | High sensitivity for detecting small transients |
Experimental Calibration Methodologies
Accurate calibration requires experimentally determining the parameters in the equations above. Two primary methodologies are used: in vitro calibration using calibration buffers and in situ calibration within the cellular environment.
1In VitroCalibration Using Buffers
This method involves creating a series of solutions with known free Ca²⁺ concentrations and measuring the indicator's fluorescence in these controlled conditions [96].
Protocol for Generating a Calibration Curve:
- Prepare Calcium Calibration Buffers: Use a reciprocal dilution method with two stock solutions: 10 mM K₂EGTA (zero Ca²⁺) and 10 mM CaEGTA (high Ca²⁺), both in a buffer containing 100 mM KCl and 30 mM MOPS at pH 7.2. Blending these stocks produces buffers with free [Ca²⁺] from 0 to 39 μM [96].
- Add Indicator: Dilute the calcium indicator into each buffer to a final concentration of 1-10 μM.
- Measure Fluorescence: Using a fluorometer or imaging system, record the fluorescence spectrum of the indicator in each buffer. For ratiometric dyes like Fura-2, collect data at both excitation wavelengths (340 nm and 380 nm) [96].
- Plot and Calculate Kd: Plot the fluorescence intensity (or ratio for ratiometric dyes) against the known [Ca²⁺]. The Kd of the indicator is the [Ca²⁺] at which half-maximal fluorescence is achieved. The data can be linearized using a double log plot of log[(F - Fmin)/(Fmax - F)] vs. log[Ca²⁺], where the x-intercept is the log(K_d) [96].
Table 2: Example Free [Ca²⁺] in Calibration Buffers at pH 7.2, 20°C
| CaEGTA (mM) | K₂EGTA (mM) | [Ca²⁺]free (μM) |
|---|---|---|
| 0.0 | 10.0 | ~0.000 |
| 1.0 | 9.0 | 0.017 |
| 2.0 | 8.0 | 0.038 |
| 5.0 | 5.0 | 0.150 |
| 8.0 | 2.0 | 0.602 |
| 9.0 | 1.0 | 1.355 |
| 10.0 | 0.0 | 39.000 |
Critical Considerations:
- The Kd of the chelator (EGTA) is highly dependent on pH, temperature, and ionic strength. A change of just 0.05 pH units can alter the Kd by up to 20% [96].
- Always perform calibrations under conditions that match your experimental setup as closely as possible.
2In SituCalibration in Live Cells
While in vitro calibration is fundamental, the intracellular environment can affect indicator performance. In situ calibration determines the key parameters Fmin, Fmax, and the effective K_d directly within the cell.
Protocol for In Situ Calibration with Single-Wavelength Indicators:
This protocol is designed for unperturbed cells, such as those loaded with dye via electroporation, where patch-clamping is not desirable [95].
- Record Resting and Active Fluorescence: Acquire a baseline recording (F_rest) and record calcium transients during physiological activity (F(t)).
- Determine Fmax at Experiment End: To find the maximum fluorescence (Fmax), perfuse the cell with a solution containing a calcium ionophore, such as ionomycin (10-20 μM), and a high extracellular [Ca²⁺] (e.g., 10 mM). This creates a saturating calcium influx, driving the indicator to its maximum fluorescence without adding significant background fluorescence from the extracellular space [95].
- Determine Fmin (Optional but Recommended): After obtaining Fmax, perfuse the cell with a Ca²⁺-free buffer containing a high concentration of a calcium chelator (e.g., 10 mM EGTA or BAPTA) to deplete intracellular Ca²⁺ and measure the minimum fluorescence (F_min).
- Calculate Absolute [Ca²⁺]: With Frest, Fmax, and Fmin known, the resting calcium concentration ([Ca²⁺]rest) and the change during activity (Δ[Ca²⁺]) can be calculated using the formula:
This method has been validated to provide accurate measurements even in small neuronal structures like dendrites and spines [95].
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for Calcium Imaging and Calibration
| Reagent / Material | Function / Purpose | Example Use Case |
|---|---|---|
| Fura-2 AM | Ratiometric, cell-permeable chemical Ca²⁺ indicator. | Agonist-induced Ca²⁺ signaling experiments in live cells [4] [39]. |
| Ionomycin | Calcium ionophore used to permeabilize cells to Ca²⁺. | Critical for obtaining F_max during in situ calibration protocols [95]. |
| EGTA & CaEGTA Stocks | Calcium buffering system for preparing calibration solutions. | Creating in vitro calibration buffers with precisely known free [Ca²⁺] [96]. |
| GCaMP8f | Genetically encoded calcium indicator (GECI) with high sensitivity. | Imaging of presynaptic Ca²⁺ dynamics at the Drosophila neuromuscular junction [97]. |
| Hank's Buffered Salt Solution (HBSS) | Physiological salt solution for maintaining cells during imaging. | Used as a base for dye loading and during live-cell imaging experiments [39]. |
| Pluronic F-127 | Non-ionic surfactant to aid dispersion of AM-ester dyes. | Facilitates loading of hydrophobic indicator dyes like Fura-2 AM into cells. |
Visualization of Workflows
The following diagrams illustrate the core logical and experimental pathways described in this guide.
Diagram 1: From Raw Fluorescence to Absolute [Ca²⁺]
This diagram outlines the core computational workflow for converting a raw fluorescence trace into a plot of absolute intracellular calcium concentration.
Diagram 2: In Situ Calibration Experimental Protocol
This diagram details the step-by-step experimental procedure for performing an in-situ calibration in live cells using ionomycin.
Advancing from relative ΔF/F0 measurements to absolute calcium concentrations is a transformative step in calcium imaging research. While ΔF/F0 is perfectly suited for reporting the timing and kinetics of calcium signals, absolute quantification is indispensable for mechanistic understanding, predictive modeling, and robust cross-study comparisons. By applying the principles of indicator binding and following rigorous calibration protocols—whether in vitro with buffers or in situ within living cells—researchers can unlock a more precise and quantitative view of the calcium code that governs cellular function. This precision is paramount for advancing fundamental research and accelerating the discovery of novel therapeutics that modulate calcium signaling pathways.
Benchmarking Performance: Sensor Validation, Comparison, and Future Directions
Calcium ions (Ca²⁺) function as a ubiquitous second messenger, regulating an extensive array of physiological processes including neurotransmission, muscle contraction, immune response, gene expression, and cell proliferation [46] [98]. The precise spatial and temporal dynamics of intracellular Ca²⁺ concentrations, termed "Ca²⁺ signatures," encode specific information that cells translate into appropriate physiological responses [46]. These signals operate over a wide dynamic range, with cytoplasmic Ca²⁺ concentrations fluctuating from approximately 100 nM at rest to micromolar levels during signaling events, while endoplasmic reticulum stores can maintain Ca²⁺ at 100-800 μM [98]. Deciphering this complex calcium code requires sophisticated detection methods capable of capturing these rapid, often subtle, concentration changes within living cells [93].
The advancement from early Ca²⁺-binding dyes to modern genetically encoded calcium indicators (GECIs) and label-free detection technologies has revolutionized our ability to monitor Ca²⁺ signaling with high spatiotemporal resolution [46] [37]. Each methodological approach presents distinct advantages and limitations across key performance metrics that directly impact experimental outcomes. This technical guide provides an in-depth analysis of these critical performance parameters—dynamic range, Kd (dissociation constant), signal-to-noise ratio (SNR), and kinetics—within the context of live-cell Ca²⁺ imaging research, offering researchers a framework for selecting appropriate methodologies for specific experimental applications.
Critical Performance Metrics in Calcium Detection
Dynamic Range
Definition and Significance: Dynamic range (DR) quantifies the maximum fluorescence change an indicator undergoes between Ca²⁺-free (Fmin) and Ca²⁺-saturated (Fmax) states, typically expressed as ΔF/Fmin or Fmax/Fmin [28]. A higher DR enables better detection of small Ca²⁺ transients and improves signal discrimination against background noise, which is particularly crucial for detecting single action potentials in neuronal imaging or elementary Ca²⁺ release events from intracellular stores [28] [6].
Comparative Performance Across Indicators: Recent developments in GECI technology have substantially improved DR values. The NEMOer series of ER-targeted GECIs demonstrates exceptional DR values ranging from 68.3 to 349.3 in cellular environments, representing a 14 to 80-fold improvement over previous standards like G-CEPIA1er (DR = 4.5) [28]. In the cytosol, jGCaMP8 sensors achieve approximately twice the sensitivity index (d') for single action potential detection compared to their predecessor jGCaMP7s, reflecting their enhanced dynamic response to neural activity [6]. For synthetic dyes, Fluo-8 offers advantages over earlier generations (Fluo-3, Fluo-4) with higher brightness and improved temperature stability, making it particularly suitable for live-cell imaging applications under physiological conditions [29].
Kd (Dissociation Constant)
Definition and Significance: The dissociation constant (Kd) represents the Ca²⁺ concentration at which half the indicator molecules are bound to Ca²⁺, defining the indicator's affinity and optimal detection range [98]. Indicators should be selected with Kd values appropriate for the expected Ca²⁺ concentrations in the cellular compartment of interest—typically low-affinity indicators (Kd > 1 μM) for high-Ca²⁺ environments like the ER, and high-affinity indicators (Kd < 1 μM) for detecting subtle cytosolic Ca²⁺ transients [28].
Indicator Affinity Ranges: The NEMOer indicators exemplify low-affinity sensors optimized for ER/SR environments, with Kd values in the near-millimolar range (comparable to the ~700 μM Kd of G-CEPIA1er) [28]. In contrast, cytosolic indicators like the jGCaMP series are engineered with significantly higher affinities (Kd < 1 μM) to detect the nanomolar to micromolar fluctuations characteristic of cytoplasmic signaling [6]. Synthetic dyes cover a broad spectrum of affinities, with Fura-2 (Kd = 0.23 μM) and OGB-1 (Kd = 0.17 μM) serving as high-affinity options, while Mag-Fura-2 (Kd = 25 μM) provides lower affinity suitable for distinguishing Ca²⁺ from Mg²⁺ [98].
Signal-to-Noise Ratio (SNR)
Definition and Significance: SNR quantifies the strength of the Ca²⁺-dependent signal relative to background noise, directly impacting the detectability of physiological events [6] [99]. Higher SNR enables more reliable detection of small transients, improves accuracy in spike detection algorithms for neuronal imaging, and enhances the fidelity of population-level analyses in complex tissues [6] [99].
Performance Comparisons: The jGCaMP8 series demonstrates significant SNR improvements, with jGCaMP8s exhibiting approximately twice the sensitivity index (d') for single action potential detection compared to previous benchmarks [6]. SNR challenges become particularly evident when comparing imaging data directly with electrophysiological recordings, as the transformation from spiking activity to Ca²⁺-dependent fluorescence introduces nonlinearities and filtering that can alter population-level analyses [99]. Label-free detection methods, such as the GaN photonic microchip platform, offer alternative approaches that circumvent limitations associated with photobleaching and indicator toxicity, potentially enabling long-term monitoring with consistent SNR [37].
Kinetics
Definition and Significance: Kinetics describe the temporal characteristics of indicator response, including rise times (response to Ca²⁺ increase) and decay times (response to Ca²⁺ decrease) [28] [6]. Rapid kinetics are essential for resolving individual signaling events in fast-spiking neurons or capturing the brief, localized Ca²⁺ transients that underlie fundamental cellular processes [28].
Performance Benchmarks: The jGCaMP8f indicator achieves remarkable kinetic performance with half-rise times (t₁/₂,rise) of approximately 2-6.6 ms in response to single action potentials, representing a threefold improvement over previous jGCaMP7f [6]. For ER Ca²⁺ monitoring, NEMOer-f displays fast dissociation kinetics (koff = 156.75 ± 3.11 s⁻¹) comparable to G-CEPIA1er, enabling detection of rapid Ca²⁺ release events with time constants under 60 ms [28]. These advancements are particularly crucial for capturing elementary signaling events such as Ca²⁺ blinks from the sarcoplasmic reticulum of cardiomyocytes, which were successfully detected for the first time using the NEMOer-f indicator [28].
Comparative Performance Tables
Table 1: Performance Metrics of Selected Synthetic Calcium Dyes
| Synthetic Dye | Class | Excitation/Emission (nm) | Kd for Ca²⁺ (μM) | Dynamic Range | Key Applications |
|---|---|---|---|---|---|
| Fura-2 | Ratiometric, dual excitation | 363, 335/512 | 0.23 | 45.7 | General cytosolic Ca²⁺ imaging |
| Indo-1 | Ratiometric, dual emission | 331/485, 510 | 0.36 | 12.9 | Flow cytometry, high-speed imaging |
| Fluo-4 | Single wavelength | 494/516 | 0.35 | ~100 | Confocal microscopy, high-throughput screening |
| Fluo-8 | Single wavelength | ~490/~514 | Similar to Fluo-4 | Higher than Fluo-4 | Live-cell imaging, room temperature loading |
| OGB-1 | Single wavelength | 488/515 | 0.17 | >5.7 | Neuronal population imaging |
| Cal-520 | Single wavelength | 492/514 | 0.32 | >0.6 (puff) | Detection of Ca²⁺ puffs and sparks |
| Mag-Fluo-4 | Single wavelength | 485/520 | 22 (4.7 mM for Mg²⁺) | 1.61 (1 AP) | Distinguishing Ca²⁺ from Mg²⁺ |
Table 2: Performance Metrics of Genetically Encoded Calcium Indicators (GECIs)
| GECI | Localization | Kd (μM) | Dynamic Range (ΔF/Fmin) | Kinetics (koff, s⁻¹) | Key Applications |
|---|---|---|---|---|---|
| jGCaMP8s | Cytosolic | <1 | High (2× d' vs jGCaMP7s) | Medium | Sensitive detection of single APs |
| jGCaMP8f | Cytosolic | <1 | Moderate | Very fast (t₁/₂,rise: 2-6.6 ms) | High-frequency neural coding |
| NEMOer-f | ER/SR | ~700 | 68.3 | 156.75 | Elementary ER/SR Ca²⁺ release events |
| NEMOer-b | ER/SR | ~700 | 139.3 | ~17-36 | Low-phototoxicity applications |
| NEMOer-c | ER/SR | ~700 | 349.3 | ~17-36 | High-contrast ER Ca²⁺ imaging |
| G-CEPIA1er | ER/SR | 706 ± 48 | 4.5 | 131.47 | Baseline ER Ca²⁺ monitoring |
Table 3: Emerging Calcium Detection Technologies
| Technology | Detection Method | Key Performance Advantages | Limitations | Applications |
|---|---|---|---|---|
| GaN Photonic Microchip | Label-free, refractive index | Sensitivity: 13,524 nA/RIU; No photobleaching | Requires specialized equipment | Long-term single-cell monitoring |
| NEMOer Series | Fluorescence (GECI) | DR up to 80× higher than previous ER sensors | Requires genetic manipulation | ER Ca²⁺ dynamics in excitable cells |
| jGCaMP8 Series | Fluorescence (GECI) | Ultra-fast kinetics (2 ms rise times) | May buffer cytosolic Ca²⁺ | Neural population imaging |
Experimental Protocols for Calcium Imaging
Live-Cell Calcium Imaging in a Microfluidic Device
This protocol outlines the process for performing live Ca²⁺ imaging on human lung endothelial cells subjected to shear stress, incorporating key performance metrics optimization [29].
Materials and Equipment:
- Human Lung Microvascular Endothelial Cells (HMVEC-Ls)
- Ibidi Luer VI microfluidic device
- EGM-2 Bulletkit culture medium
- Fluo-8 Calcium Flux Assay Kit
- Ismatec REGLO ICC Digital peristaltic pump
- Lionheart FX automated microscope or equivalent with 20× objective
- Temperature-controlled incubation chamber
Procedure:
- Cell Culture Preparation: Maintain HMVEC-Ls in T-25 or T-75 flasks using complete EGM-2 medium, replacing medium daily. Use cells between passages 3-7 at 70-90% confluency for experiments [29].
- Microfluidic Device Setup: Assemble and sterilize the flow system, including inlet tubing (5.5 cm of 1/8" ID tubing connected to 22.5 cm of 1/16" ID tubing) and outlet tubing (4.5 cm of 1/8" ID tubing connected to 22 cm of 1/16" ID tubing) using autoclave sterilization [29].
- Cell Loading and Seeding: Harvest cells using TrypLE Express without phenol red and neutralize with trypsin neutralizing solution. Seed cells into the microfluidic channel at appropriate density and incubate for 24-48 hours to reach desired confluency [29].
- Calcium Indicator Loading: Prepare Fluo-8 working solution in HBSS according to manufacturer instructions. Load Fluo-8 into cells by incubating for 30-60 minutes at room temperature, taking advantage of Fluo-8's temperature stability [29].
- Shear Stress Application: Establish flow using the peristaltic pump to generate defined shear stress conditions relevant to vascular physiology (typically 5-20 dyn/cm²) [29].
- Image Acquisition: Perform time-lapse imaging using appropriate filter sets for Fluo-8 (Ex/Em ~490/514 nm). Acquire images at frequencies sufficient to capture expected Ca²⁺ transients (typically 0.5-10 Hz depending on application) [29].
- Data Analysis: Process time-lapse sequences using imaging software (e.g., ImageJ with LC Pro Plugin). Calculate ΔF/F0 values where F0 represents baseline fluorescence before stimulation. Identify Ca²⁺ transients using threshold-based detection algorithms [29].
Protocol for Evaluating GECI Performance in Neuronal Cultures
This protocol describes methodology for characterizing GECI performance metrics in primary neuronal cultures, particularly focusing on kinetic parameters and SNR [6].
Materials and Equipment:
- Primary neuronal cultures
- jGCaMP8 or other GECI constructs (viral or transfection delivery)
- Field stimulation apparatus for action potential induction
- High-speed imaging system with appropriate magnification
- Analysis software with spike inference capabilities
Procedure:
- GECI Expression: Introduce GECI construct into neurons using appropriate method (lentiviral transduction, calcium phosphate transfection, or electroporation). Allow 7-14 days for sufficient expression, balancing signal intensity with potential cytotoxicity [6].
- Action Potential Evocation: Use field stimulation to elicit defined action potential trains (single spikes to high-frequency bursts) while maintaining physiological temperature and conditions [6].
- High-Speed Imaging: Acquire fluorescence images at high temporal resolution (≥100 Hz) to adequately sample the rapid fluorescence transients. Use appropriate illumination intensities to maximize SNR while minimizing photobleaching and phototoxicity [6].
- Simultaneous Electrophysiology (Optional): Perform simultaneous patch-clamp recording and calcium imaging in a subset of experiments to directly correlate electrical events with fluorescence transients for precise kinetic characterization [99].
- Data Analysis: Extract fluorescence traces from regions of interest corresponding to neuronal somata or processes. Calculate ΔF/F0, response kinetics (rise and decay times), and SNR. For single action potential responses, compute sensitivity index (d') to compare GECI performance across variants [6].
Diagram 1: Calcium Signaling and Detection Workflow. This diagram illustrates the fundamental pathway of calcium signaling from stimulus to cellular response, coupled with the methodological approach for detecting and analyzing these signals through appropriate indicator selection and imaging strategies.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 4: Key Research Reagent Solutions for Calcium Imaging
| Category | Specific Examples | Function/Application | Performance Considerations |
|---|---|---|---|
| Synthetic Dyes | Fluo-8, Fura-2, Indo-1, Cal-520 | Chemical indicators loaded into cells | Fluo-8 offers higher brightness and temperature stability than Fluo-3/4 [29] |
| GECIs (Cytosolic) | jGCaMP8 series (8s, 8f, 8m) | Genetically encoded indicators for cytosolic Ca²⁺ | jGCaMP8f: ultra-fast kinetics (2 ms rise time); jGCaMP8s: highest sensitivity [6] |
| GECIs (Organellar) | NEMOer series (f, m, c, b, s) | ER/SR-targeted Ca²⁺ indicators | Dynamic ranges 14-80× higher than previous ER sensors [28] |
| Microfluidic Devices | Ibidi Luer VI | Applying controlled shear stress during imaging | Enables physiological flow conditions during live-cell imaging [29] |
| Flow Systems | Ismatec REGLO peristaltic pump | Maintaining continuous media flow | Cost-effective with lower contamination risk vs. syringe pumps [29] |
| Imaging Systems | Lionheart FX microscope | Automated time-lapse imaging | Requires 20× objective and environmental control [29] |
| Analysis Software | ImageJ with LC Pro Plugin | Quantifying calcium transients from time-lapse data | Enables calculation of ΔF/F₀ and kinetic parameters [29] |
Advanced Detection Technologies
Label-Free Detection Methods
Emerging technologies that monitor Ca²⁺ dynamics without fluorescent indicators address several limitations inherent to both synthetic dyes and GECIs. The GaN (gallium nitride) photonic microchip represents a promising approach that detects refractive index variations in single cells induced by Ca²⁺ signaling [37]. This platform achieves a sensitivity of 13,524 nA/RIU and enables scalable, parallel monitoring of multiple single cells, revealing cell-to-cell heterogeneity in Ca²⁺ response kinetics without the complications of photobleaching or indicator toxicity [37]. The system integrates a complete working prototype with portable electronics and smartphone connectivity, facilitating potential applications in remote monitoring and point-of-care diagnostics [37].
Synthetic Biology Approaches
Synthetic biology principles are being applied to overcome persistent challenges in Ca²⁺ detection optimization [98]. By employing standardized, modular biological components ("parts"), these approaches aim to enhance the reliability of controlling cellular behavior and improve the precision of Ca²⁺ signaling measurement [98]. Engineering efforts focus on refining the interface between endogenous Ca²⁺ signaling pathways and synthetic reporter systems, potentially leading to next-generation indicators with improved performance characteristics and reduced interference with native cellular processes [98].
Diagram 2: Calcium Detection Technology Selection Guide. This decision-support diagram illustrates the relationship between experimental requirements, performance metric priorities, and recommended detection technologies, helping researchers select optimal methodologies for specific applications.
The quantitative performance metrics of dynamic range, Kd, signal-to-noise ratio, and kinetics form the fundamental framework for evaluating and selecting calcium detection methodologies in live-cell research. Recent advancements in both fluorescent indicator technology and label-free detection platforms have substantially improved these parameters, enabling researchers to address increasingly complex biological questions with greater temporal and spatial precision. The development of GECIs with dramatically expanded dynamic ranges (NEMOer series) and accelerated kinetics (jGCaMP8 series) has opened new possibilities for capturing elementary calcium signaling events that were previously undetectable. Concurrently, emerging label-free technologies offer complementary approaches for long-term monitoring without the limitations of photobleaching or indicator toxicity. As these technologies continue to evolve, guided by synthetic biology principles and engineering insights, they will further enhance our ability to decipher the complex calcium code underlying fundamental physiological processes and pathological conditions, ultimately accelerating drug discovery and therapeutic development across a broad spectrum of human diseases.
Calcium ions (Ca²⁺) function as ubiquitous intracellular second messengers, regulating processes from synaptic transmission and muscle contraction to gene expression and cell death [100]. The ability to monitor calcium dynamics in living cells is therefore crucial for understanding cellular communication and signaling pathways. Genetically Encoded Calcium Indicators (GECIs) represent a cornerstone technology for real-time monitoring of intracellular Ca²⁺ signals in living organisms [101]. Unlike synthetic dyes that require invasive loading procedures and lack cell-type specificity, GECIs can be genetically targeted to specific cell populations using tissue-specific promoters, enabling long-term monitoring of calcium dynamics in intact biological systems [100].
The GCaMP family, first developed in 2001 by Junichi Nakai, has emerged as one of the most widely adopted GECI platforms [100] [102]. These indicators are synthetic fusion proteins consisting of a circularly permuted green fluorescent protein (cpGFP), calmodulin (CaM), and the M13 peptide sequence derived from myosin light-chain kinase [102]. In the absence of calcium, the GCaMP chromophore exists in a protonated state with minimal fluorescence. When calcium binds to the calmodulin domain, it triggers a conformational change where CaM wraps around the M13 peptide, protecting the chromophore from solvent access and promoting its deprotonation. This transition to an anionic state results in a significant increase in green fluorescence, with excitation and emission peaks at approximately 480 nm and 510 nm, respectively [100] [102]. This molecular mechanism enables researchers to quantify neuronal activity, track signaling pathway activation, and study calcium-mediated processes in everything from cardiac conduction to developmental biology [100].
Molecular Engineering and Design Principles
Structural Scaffolds and Optimization Strategies
The continuous evolution of GECIs has been driven by systematic engineering of their core components. The GCaMP scaffold has undergone iterative improvements through multiple generations, from GCaMP1 to the recent jGCaMP8 series [100] [102]. Each generation has addressed limitations in brightness, kinetics, sensitivity, or biocompatibility through structure-guided mutagenesis and high-throughput screening. The jGCaMP8 series, developed through large-scale screening of over 1,000 mutations, represents a significant advancement by replacing the traditional RS20 peptide with a novel peptide from endothelial nitric oxide synthase (ENOSP), which dramatically improves kinetics without compromising sensitivity [103] [104].
Alternative engineering approaches have yielded distinct sensor families. The XCaMP indicators utilize a different calmodulin-binding peptide from CaM-dependent kinase kinase (ckkap), providing faster kinetics than previous GCaMP versions [103]. Meanwhile, the NEMO sensors represent a fundamental architectural innovation by combining the GCaMP and NCaMP7 design strategies with the bright monomeric green fluorescent protein mNeonGreen (mNG) as the fluorophore [101]. This hybrid approach leverages the brightness of mNG while maintaining calcium-dependent fluorescence changes, resulting in dramatically improved dynamic ranges exceeding 100-fold in some variants [101].
Key Engineering Innovations
The development of jGCaMP8 involved optimizing three critical regions: the calmodulin-binding peptide, the linkers connecting the domains, and the interface between calmodulin and M13 [103] [104]. By systematically testing 30 different peptides to replace RS20, followed by extensive mutagenesis of linkers and interface residues, researchers achieved a sensor with both ultra-fast kinetics and high sensitivity to neural activity [104]. The crystal structure of the leading variant (jGCaMP8.410.80) guided further optimization through site-saturation mutagenesis, particularly targeting residues near the ENOSP C-terminus and the domain interface [103].
The NEMO sensors were engineered using a different strategy, applying known GCaMP design principles to mNeonGreen and screening for constructs with high dynamic range and low basal fluorescence [101]. The most successful NEMO variants adopted an NCaMP7-like design where the CaM-M13 sensing module is inserted into the middle of the fluorescent protein rather than at the termini [101]. This architectural innovation, combined with the intrinsic brightness of mNeonGreen, enables NEMO sensors to achieve peak signal-to-baseline ratios approximately 20-fold larger than the GCaMP6 series [101].
Figure 1: Structural Design Principles of Leading GECI Families. Each sensor family employs distinct engineering strategies: GCaMP/jGCaMP8 uses terminal M13 and CaM domains with cpEGFP; XCaMP incorporates the CKKAP peptide for faster kinetics; NEMO utilizes mNeonGreen with an internal CaM-M13 insertion for enhanced dynamic range.
Technical Performance Comparison
Biophysical Properties
The quantitative performance of GECIs varies significantly across different variants, with specific trade-offs between sensitivity, kinetics, and dynamic range. The following table summarizes the key biophysical parameters for the major GECI families:
Table 1: Biophysical Properties of Leading GECI Variants
| Sensor | Variant | Kd (nM) | Hill Coefficient | Dynamic Range (ΔF/F) | Half-Rise Time (ms) | Half-Decay Time (ms) |
|---|---|---|---|---|---|---|
| jGCaMP8 | 8f (fast) | 334 ± 18 | 2.08 ± 0.22 | 0.41 ± 0.12 | 7.1 ± 0.74 | 67.4 ± 11.2 |
| 8m (medium) | 108 ± 3 | 1.92 ± 0.12 | 0.76 ± 0.22 | 7.1 ± 0.61 | 118.3 ± 13.2 | |
| 8s (sensitive) | 46 ± 1 | 2.20 ± 0.13 | 1.11 ± 0.22 | 10.1 ± 0.86 | 306.7 ± 32.2 | |
| GCaMP7 | 7f (fast) | 150 ± 2 | 3.10 ± 0.16 | 0.21 ± 0.1 | 24.8 ± 6.6 | 181.9 ± 76.0 |
| NEMO | NEMOf | Not reported | Not reported | 194.3 ± 7.7* | Not reported | Not reported |
| NEMOc | Not reported | Not reported | 112.0 ± 9.8* | Not reported | Not reported | |
| NEMOm | Not reported | Not reported | 101.9 ± 6.6* | Not reported | Not reported |
Note: NEMO dynamic range values represent peak signal-to-baseline ratio (ΔF/F₀) in response to carbachol stimulation in HEK293 cells, measured differently than the purified protein assays used for GCaMP variants [101].
The jGCaMP8 series demonstrates remarkable improvements over previous generations, with jGCaMP8f showing a 4-fold faster rise time and 2.5-fold faster decay time compared to jGCaMP7f [105]. Meanwhile, jGCaMP8s is twice as sensitive as jGCaMP7s and more than twice as fast as jGCaMP7f at detecting single action potentials [105]. The NEMO sensors exhibit extraordinary dynamic ranges, with NEMOf showing a 194-fold peak signal-to-baseline ratio in response to muscarinic receptor activation in HEK293 cells, approximately 25 times higher than GCaMP6m under the same conditions [101].
Performance in Biological Systems
When deployed in actual experimental systems, each sensor family demonstrates distinct strengths. The jGCaMP8 sensors have been extensively validated in both Drosophila and mouse models, showing superior performance for detecting neural activity in vivo [105]. In Drosophila neuromuscular junctions, jGCaMP8f and jGCaMP8m responses at 10 Hz and 20 Hz stimulation were "dramatically more pronounced than jGCaMP7f" [105]. Similarly, in mouse visual cortex, jGCaMP8 sensors enabled tracking of large populations of neurons on timescales relevant to neural computation, with half-rise times as fast as 2 milliseconds [103] [104].
The NEMO sensors excel in scenarios requiring detection of subtle calcium transients or operation in high-background environments. When reporting carbachol-induced calcium transients in HEK293 cells, NEMOf achieved a peak signal-to-baseline ratio of 194.3 ± 7.7, compared to approximately 8 for GCaMP6m and jGCaMP8f under identical conditions [101]. This exceptional contrast ratio enables NEMO sensors to detect weak calcium signals that would be challenging to resolve with other indicators.
Table 2: Experimental Performance Comparison in Different Model Systems
| Application Context | Recommended Sensor | Performance Advantages | Key Supporting Evidence |
|---|---|---|---|
| High-frequency neural coding | jGCaMP8f | Ultra-fast kinetics (2 ms half-rise time) | Tracks individual spikes at up to 50 Hz in mouse cortex [103] |
| Detection of single action potentials | jGCaMP8s | Highest sensitivity for sparse activity | 2x higher sensitivity than jGCaMP7s for 1 AP detection [105] |
| Weak signal detection in nonexcitable cells | NEMOf | Extreme dynamic range (>100-fold) | 25x higher ΔF/F than GCaMP6m in HEK293 cells [101] |
| Long-term chronic imaging | GCaMP-X | Reduced cytotoxicity | Maintains healthy neurite morphology over weeks [67] |
| Drosophila synaptic imaging | jGCaMP8m | Balanced speed and sensitivity | Dramatically improved bouton responses vs jGCaMP7f [105] |
Experimental Protocols and Methodologies
Sensor Expression and Delivery
Effective deployment of GECIs requires careful consideration of expression strategies. Plasmid vectors for jGCaMP8 sensors are available from Addgene, with adeno-associated virus (AAV) serotypes suitable for in vivo delivery in multiple model organisms [105] [106]. For mammalian systems, the synapsin promoter (syn) provides neuron-specific expression, while the CAG or EF1α promoters enable broader expression across cell types [105]. Transgenic mice expressing jGCaMP8 variants are available through JAX Laboratories, and jGCaMP8-expressing flies can be obtained from the Bloomington Drosophila Stock Center [105].
For cellular imaging, transfection with Lipofectamine 2000 or similar reagents typically achieves sufficient expression within 24-48 hours. For more challenging preparations such as primary neuronal cultures, lentiviral delivery often provides higher transduction efficiency and more consistent expression levels. The NEMO sensors have been successfully expressed in HEK293 cells using standard transfection protocols, with performance validated using a P2A-based bicistronic vector coexpressing mKate as an expression marker [101].
Calcium Imaging Workflow
A standardized imaging protocol for GECI experiments includes the following key steps:
Sample Preparation: Culture cells or prepare tissue slices expressing the GECI of interest. For in vivo imaging, prepare animal surgery following institutional guidelines.
System Calibration: Perform background subtraction using regions without sensor expression. Adjust laser power or illumination intensity to avoid sensor saturation while maintaining sufficient signal-to-noise ratio.
Data Acquisition: Acquire time-series images at appropriate sampling rates. For jGCaMP8 sensors detecting fast neuronal spikes, frame rates of 50-100 Hz are recommended [103]. For slower calcium oscillations in nonexcitable cells, 1-10 Hz may be sufficient.
Stimulation Paradigm: Apply appropriate stimuli based on the biological question. For neuronal cultures, electrical field stimulation can evoke action potentials [103]. For receptor signaling studies, apply ligands such as carbachol (10 μM) or ATP [101] [107].
Data Analysis: Extract fluorescence traces (F) from regions of interest and calculate ΔF/F₀, where F₀ represents baseline fluorescence. For spike detection in neuronal preparations, use deconvolution algorithms optimized for the specific sensor kinetics [103].
Figure 2: Generalized Workflow for GECI Experiments. The protocol begins with sensor selection based on experimental requirements, followed by appropriate expression system deployment, careful sample preparation, optimized imaging setup, controlled stimulation, systematic data acquisition, and rigorous analysis to quantify calcium transients.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for GECI Experiments
| Reagent / Resource | Function | Example Sources |
|---|---|---|
| jGCaMP8 plasmids | Sensor expression in multiple systems | Addgene [105] [106] |
| AAV-syn-jGCaMP8 | Neuron-specific in vivo expression | Addgene (multiple serotypes) [105] |
| Transgenic mice | Stable expression without viral delivery | JAX Laboratories [105] |
| jGCaMP8 Drosophila | Insect neural circuit imaging | Bloomington Drosophila Stock Center [105] |
| P2A-mKate vectors | Expression normalization control | Literature constructs [101] |
| Ionomycin & Thapsigargin | Calcium store manipulation for range quantification | Commercial suppliers [101] |
| Carbamylcholine (CCh) | Muscarinic receptor activation | Commercial suppliers [101] |
| ATP | Purinergic receptor stimulation | Commercial suppliers [107] |
Applications in Calcium Signaling Research
Neuronal Activity Monitoring
GCaMP sensors have revolutionized neuroscience by enabling large-scale recording of neural activity in behaving animals [100]. The jGCaMP8 series represents a particular advancement for studying high-frequency neural coding. With rise times as fast as 2 milliseconds, these sensors can resolve individual action potentials even during rapid bursts of activity up to 50 Hz [103] [104]. This temporal resolution begins to approach the timescales of actual neural computation, allowing researchers to track information flow through neural circuits with unprecedented precision [104]. In mouse visual cortex, jGCaMP8 sensors have enabled researchers to follow the millisecond-scale dynamics of sensory processing, revealing how individual neurons integrate visual information [105] [103].
Signaling Pathway Analysis
Beyond neuronal activity, GECIs provide powerful tools for dissecting calcium-mediated signaling pathways in diverse cell types. The exceptional dynamic range of NEMO sensors makes them particularly valuable for monitoring subtle calcium transients in nonexcitable cells [101]. When expressed in HEK293 cells, NEMO sensors detect carbachol-induced muscarinic receptor activation with signal-to-baseline ratios exceeding 100-fold, enabling precise quantification of GPCR signaling dynamics [101]. Similarly, in endothelial cells (HUVEC), modified jGCaMP8f variants with improved dynamic range successfully monitor ATP-induced calcium signaling, demonstrating utility for vascular biology research [107].
Long-term Imaging and Chronic Studies
For longitudinal studies requiring extended sensor expression, the GCaMP-X platform provides reduced cytotoxicity compared to conventional GCaMP indicators [67]. By incorporating an additional calmodulin-binding motif, GCaMP-X minimizes interference with endogenous calcium signaling pathways, preserving normal neuronal morphology and function over weeks of expression [67]. In cortical neurons, GCaMP-X expression for one month or longer enabled chronic monitoring of spontaneous calcium transients as they developed into autonomous global oscillations, revealing calcium dynamics during neurite outgrowth and network maturation [67].
The ongoing development of GECIs continues to expand the frontiers of calcium imaging research. The jGCaMP8 series sets a new standard for speed and sensitivity in green indicators, while the NEMO family demonstrates the potential for dramatically improved dynamic range through innovative scaffold engineering [103] [101]. Looking forward, several promising directions emerge for further advancement. Red and far-red indicators continue to improve, enabling deeper tissue penetration and multicolor imaging when combined with green sensors [100]. Reduced cytotoxicity platforms like GCaMP-X facilitate long-term studies of neural development and plasticity [67]. Additionally, continued optimization of sensor kinetics will further narrow the gap between calcium imaging and the millisecond-timescale of electrical signaling in neurons.
For researchers selecting GECIs for specific applications, the optimal choice depends heavily on experimental priorities. The jGCaMP8 series currently offers the best combination of speed and sensitivity for most neuronal imaging applications, with variant selection (8f, 8m, or 8s) dictated by the specific balance of temporal resolution and detection sensitivity required [105] [103]. For applications demanding maximum signal contrast or detection of subtle calcium transients, particularly in nonexcitable cells, NEMO sensors provide unprecedented dynamic range [101]. When planning long-term experiments requiring extended sensor expression, GCaMP-X variants minimize cellular perturbations while maintaining robust calcium detection [67].
As calcium imaging continues to illuminate diverse biological processes from neural computation to cellular signaling, these advanced GECI tools provide researchers with increasingly powerful means to visualize and quantify calcium dynamics in living systems. The complementary strengths of different sensor families enable experimental designs tailored to specific biological questions, driving continued discovery in the fundamental mechanisms of calcium signaling across physiological and pathological contexts.
In the field of live-cell research, calcium signaling detection is fundamental to understanding a vast array of physiological processes, from neurotransmitter release to cell proliferation and gene expression. The development of quantitative kinetic models is crucial for deciphering the complex dynamics of these signaling pathways. However, a significant challenge persists: many published models are fit to different data sets, often without rigorous cross-validation, and sometimes with parameters that are underdetermined due to being tuned as part of a larger reaction cascade [108]. This lack of standardization makes it difficult to compare models and trust their predictive power outside specific experimental conditions.
Non-linear mixed effects (NLME) modeling emerges as a powerful statistical framework to address these limitations. This guide provides an in-depth technical overview of how NLME modeling can be applied for the rigorous validation of kinetic schemes, with a specific focus on calcium signaling pathways. By explicitly accounting for multiple sources of variability, NLME offers a more robust approach to parameter estimation and model selection, ultimately leading to more reliable and generalizable biological models.
Theoretical Foundations of NLME for Kinetic Modeling
Non-linear mixed effects modeling is a hierarchical framework designed to handle data where multiple measurements are taken from the same experimental units (e.g., repeated calcium imaging from the same cell culture or subject). Its power lies in its ability to disentangle different sources of variability.
Core Components of the NLME Framework
- Fixed Effects: These parameters represent the average, typical kinetics of the population. For a calcium-calmodulin binding model, the fixed effects would describe the average reaction rate constants for the cohort being studied.
- Random Effects: These quantify the subject-specific or experiment-specific deviations from the population average. They account for the variability between individuals (e.g., variations in protein expression levels between cell cultures) that cannot be explained by the fixed effects alone.
- Residual Error: This captures the unexplained, random variability within individuals, such as measurement noise or minor fluctuations in experimental conditions during a single time-series acquisition.
The application of NLME is particularly potent in experimental contexts where a key variable cannot be controlled precisely. For instance, in calcium imaging experiments involving laser-induced uncaging, the exact amount of calcium released can be uncertain and may vary from trial to trial. NLME modeling can incorporate this uncertainty as a random effect, allowing for the accurate estimation of the underlying kinetic parameters despite the technical variability [108].
Case Study: Application to Calcium-Calmodulin Kinetic Schemes
A recent study by Linkevicius et al. (2025) provides a paradigm for the application of NLME modeling to calcium signaling kinetics [108]. The study addressed critical limitations in existing calmodulin models by fitting multiple published kinetic schemes to a common benchmark data set.
Experimental Data Integration
The common data set combined two complementary types of information:
- Equilibrium Data: From Shifman et al., providing information on the steady-state binding properties.
- Dynamic Data: From Faas et al., consisting of fluorescence time-series measurements following laser-induced Ca²⁺ uncaging, which informs on the temporal dynamics of the binding process [108].
The technical uncertainty in the amount of uncaged calcium in the dynamic data set was a key challenge, which was effectively handled using the NLME framework.
Computational Methodology and Workflow
The analysis was implemented using the Pumas.jl package in Julia, a high-performance programming language well-suited for computational biology [108]. The following workflow outlines the key steps in the model fitting and validation process:
Key Findings and Model Comparison
The study demonstrated that parameters refit using NLME modeling provided a significantly better fit to the common data set than the originally published parameters, as quantified by lower Akaike Information Criterion (AIC) values [108]. This indicates that the published parameters were suboptimal, likely due to the limitations of their original fitting procedures.
Furthermore, the rigorous comparison revealed that a kinetic scheme featuring independent calmodulin lobes with unique, non-identical binding sites provided the best fit to the data [108]. This supports the biological hypotheses that partially bound calmodulin is significant in cellular signaling and that calcium binding sites within a lobe are kinetically distinct.
Experimental Protocols for Calcium Signaling Detection
The validity of any kinetic model is contingent on the quality of the underlying experimental data. Below are detailed protocols for key calcium detection methods relevant to generating data for kinetic modeling.
Live-Cell Calcium Imaging in 3D Cultures
This protocol, adapted from von Molitor et al. (2020), is designed for monitoring calcium signaling in complex, physiologically relevant 3D models [109].
- Cell Culture and Preparation: Generate spheroids from relevant cell lines (e.g., human tongue-derived HTC-8 cells). Transfer spheroids to a specialized imaging chamber designed for perfusion and stabilization to prevent movement artifacts during recording.
- Fluorescent Sensor Loading: Incubate spheroids with a genetically encoded or chemical Ca²⁺ indicator (e.g., G-GECO or Calbryte 520-AM). For chemical dyes, use 4 µM dye concentration in normal bath solution at 37°C for 30 minutes [109] [110].
- Microscopy and Perfusion Setup:
- Stabilization: Use custom-built or commercial chambers that securely hold the spheroid under constant perfusion.
- Imaging: Employ confocal or light sheet fluorescence microscopy. Light sheet microscopy is advantageous for larger samples, allowing acquisition over a z-depth of 100 µm without sacrificing spatial or temporal resolution [109].
- Perfusion: Apply gustatory substances (e.g., Salicine, Saccharin) or other stimuli (e.g., ATP) via a perfusion system. For bitter compounds like Saccharin, note that dose-dependent responses may require extracellular Ca²⁺ [109].
- Data Acquisition: Record time-lapse images with high spatio-temporal resolution. For example, use a 100 ms exposure time to capture rapid calcium transients [110].
- Analysis: Analyze the amplitude, latency, and propagation of Ca²⁺ signals. Signals typically decrease in amplitude and are progressively delayed from the spheroid border towards the center [109].
Programmable Microfluidic Platform for Microglia (CAM-μTAS)
This protocol leverages microfluidic technology to create biomimetic cytokine gradients for studying microglial calcium dynamics during inflammation [110].
- Device Fabrication: Fabricate the CAM-μTAS using standard photolithography and soft lithography with polydimethylsiloxane (PDMS). The device integrates Quake valves and lifting-gate microvalve arrays for precise fluidic control [110].
- Cell Culture and Seeding: Culture BV2 microglia cells. Seed cells into the microfluidic device's culture chamber at a density of 2×10⁵ cells/mL. Allow cells to adhere overnight.
- Calcium Indicator Incubation: Perfuse the device with Calbryte 520-AM (4 µM) to load the indicator into the cells. The microsystem automates this incubation and subsequent washing steps.
- Gradient Generation and Stimulation: Use the integrated pneumatic valves to generate defined concentration gradients of an "inflammatory soup" (e.g., IL-1β, IL-6, TNF-α, and ATP). The system can perform rapid media changes in 1.5 seconds [110].
- Image Acquisition and Analysis: Acquire time-lapse images using an inverted microscope (e.g., 40X magnification, 100 ms exposure). Analyze location-dependent activation patterns by quantifying parameters like calcium transient latency to peak [110].
Research Reagent and Tool Solutions
The following table details key reagents, tools, and software essential for conducting rigorous calcium signaling experiments and subsequent NLME modeling.
Table 1: Essential Research Toolkit for Calcium Signaling and NLME Modeling
| Item Name | Type/Category | Primary Function in Research |
|---|---|---|
| G-GECO / Calbryte 520-AM | Genetically Encoded / Chemical Ca²⁺ Indicator | Fluorescent reporting of changes in intracellular Ca²⁺ concentration ([Ca²⁺]ᵢ) [109] [110]. |
| CAM-μTAS | Microfluidic Device | Automates cell culture, perfusion, and application of biomimetic cytokine gradients for highly controlled calcium imaging [110]. |
| Pumas.jl | Software Package | High-performance computing environment for implementing NLME modeling, pharmacokinetic/pharmacodynamic analysis, and model validation [108]. |
| Inflammatory Soup | Experimental Stimulus | A mixture of pro-inflammatory cytokines (IL-1β, IL-6, TNF-α) and ATP used to activate immune cells like microglia and evoke calcium transients [110]. |
| Light Sheet Microscopy | Imaging Technique | Enables high-resolution, high-speed live-cell imaging of calcium dynamics in large 3D samples with minimal photobleaching [109]. |
| EMC² Algorithm | Analysis Software | Robustly tracks the position of individual neurons in calcium imaging movies from behaving animals, compensating for motion and intermittent signal detectability [111]. |
Implementation Guide for NLME Modeling
Transitioning from traditional modeling approaches to NLME requires careful consideration of data structure and software choices.
Data Preparation and Model Specification
- Data Structure: Your dataset must be in a "long" format where each row represents a single observation at a specific time for a specific experimental subject (e.g., a cell culture, a spheroid, an animal). Essential columns include: Subject ID, Time, Observed Value (e.g., fluorescence), and any covariates (e.g., stimulus type).
- Model Definition: You must define your underlying kinetic model (e.g., a system of ordinary differential equations representing calmodulin binding states) and specify which parameters are considered fixed effects and which will have random effects. The choice of random effects structure (e.g., which parameters vary between subjects) is a critical, hypothesis-driven decision.
Software and Computational Tools
While the featured case study used Pumas.jl [108], other software platforms like nlme and lme4 in R, or NONMEM, are also widely used in the life sciences. The selection of software often depends on the model's complexity, the size of the dataset, and the need for computational speed. For complex biological systems, Julia's performance advantages are becoming increasingly attractive.
The integration of non-linear mixed effects modeling with high-quality experimental data from advanced calcium detection methods represents a significant leap forward in the rigorous validation of kinetic schemes. This approach moves beyond simply finding a "best-fit" for a single data set and instead focuses on identifying models and parameters that are robust, generalizable, and account for the inherent biological and technical variability in live-cell research. By adopting this framework, researchers in drug development and basic science can build more trustworthy models of calcium signaling pathways, enhancing the predictive power of in silico experiments and accelerating the discovery of novel therapeutic strategies.
Calcium signaling is a ubiquitous and ancient intracellular and intercellular messaging system, regulating an extensive array of physiological processes from neurotransmission and muscle contraction to cell proliferation, immune responses, and embryonic development [35] [93]. The visualization and quantification of calcium dynamics in living cells, known as calcium imaging, has therefore become a cornerstone technique in biological research and drug discovery. However, the field is characterized by a profound lack of standardization. The analysis of calcium imaging data employs a wide array of methods for signal processing, baseline definition, and activity thresholding, which dramatically impacts the results and conclusions of a study [35] [93]. This methodological diversity makes comparative analysis across studies nearly impossible and threatens the reproducibility of scientific findings. This whitepaper articulates the pressing need for common data sets and standardized analysis pipelines in calcium imaging research. By framing the problem within the context of a broader thesis on detection methods, we aim to provide researchers and drug development professionals with a clear understanding of the current challenges, available tools, and a concrete path toward a more unified and robust analytical framework.
The Standardization Challenge: A Fragmented Analytical Landscape
The Core Analytical Pipeline and Its Variability
The general pipeline for analyzing calcium imaging data at cellular resolution involves several consistent steps: image processing (motion correction, background correction, region-of-interest (ROI) identification), tracking cells over time to obtain calcium time series, quantification of calcium activity, and spatiotemporal pattern analysis [35] [93]. The critical challenge is that each step is highly modular and implemented differently across labs and studies. For instance, a seminal review highlights that techniques for defining background signal and baseline measurements can drastically alter the interpretation of calcium transients [93]. Furthermore, the criteria for identifying a meaningful calcium event—the activity threshold—vary extensively. This is particularly problematic when studying systems with irregular and complex calcium activity, such as in embryonic neurons, where the lack of clear periodicity makes consistent thresholding difficult [35]. This inconsistency means that two studies analyzing seemingly similar experimental data can arrive at dramatically different conclusions based solely on their chosen analytical parameters [35] [93].
The Scalability Problem
The ever-increasing size of calcium imaging datasets, capable of monitoring large neural populations across weeks, necessitates scalable, reproducible, and fully automated analysis pipelines [112]. The common reliance on custom-written scripts, while flexible, presents a significant barrier to reproducibility and standardization. As noted in one study, "no current off-the-shelf software tool meets all... needs" of a particular lab, leading researchers to develop their own solutions, such as collections of Jupyter notebooks for analysis [4]. While these custom tools automate repetitive steps and offer diagnostic interactive data exploration, they are often tailored to a specific lab's requirements and are not universally adopted, thereby exacerbating the fragmentation of the field.
Current Software Tools and Their Capabilities
In response to these challenges, several software tools have been developed to streamline parts of the analysis workflow. The table below summarizes key available pipelines and their primary functions.
Table 1: Software Tools for Calcium Imaging Data Analysis
| Tool Name | Primary Function | Key Features | Language/Platform |
|---|---|---|---|
| Suite2p, CaImAn [14] | Pre-processing & ROI extraction | Automated processing of large datasets; motion correction; source extraction. | Python (CaImAn), MATLAB (Suite2p) |
| Jupyter Notebooks (Custom) [4] | Flexible analysis scripting | ROI detection; peak analysis; frequency analysis; movie making. | Python |
| CalciumNetExploreR (CNER) [14] | Downstream network analysis | Normalization; binarization; network construction & topology analysis. | R |
| Fiji/ImageJ Plugins (e.g., TurboReg) [14] | Image pre-processing | Motion correction; template matching for signal extraction. | Java |
These tools highlight a division of labor between pre-processing (e.g., Suite2p, CaImAn) and downstream, higher-level analysis (e.g., CNER). CNER, for instance, is an R package designed to integrate normalization, binarization, population activity visualization, network construction, and graph theory-based topological analysis into a single, cohesive pipeline [14]. This approach helps standardize the later stages of analysis, allowing for the comparison of network metrics like clustering coefficients and global efficiency.
A Pathway to Standardization: Proposing Common Workflows
A Generalized Standardized Workflow for Calcium Imaging Analysis
To address the current fragmentation, we propose the adoption of a generalized, modular workflow that can serve as a template for the community. The following diagram outlines this cohesive pathway, from data acquisition to advanced network analysis.
Detailed Methodologies for Key Analytical Steps
The workflow above encompasses several critical steps that require standardized definitions and algorithms.
A. Data Pre-processing and Normalization
The initial step involves converting raw fluorescence (F) into a normalized signal (ΔF/F) to account for heterogeneity in baseline indicator expression. A common method is min-max scaling:
x'_t = (x_t - min(x)) / (max(x) - min(x))
where x'_t is the normalized signal at time t, and min(x) and max(x) are the minimum and maximum signals over the recording [14]. This scales each cell's signal to a range between 0 and 1, enabling comparability across cells and experiments.
B. Signal Binarization and Event Detection
To distinguish active from inactive states, normalized signals are often binarized. A typical thresholding method defines a cell as active at time t if its normalized signal exceeds two standard deviations above the mean:
x'_{t,bin} = 1, if x'_t > μ_{x'} + 2σ_{x'}
x'_{t,bin} = 0, otherwise [14]
This approach identifies significant calcium transients relative to the cell's baseline activity, suppressing minor fluctuations and emphasizing meaningful events, which is crucial for subsequent correlation-based network analyses.
C. Functional Connectivity and Network Analysis
Functional connectivity is frequently assessed using pairwise cross-correlation of normalized calcium signals. The ccf() function in R (or its equivalent in Python) calculates the cross-correlation coefficient ρ_{ij}(τ) between the time series of cell i and cell j at various time lags (τ) [14]. A correlation matrix is then constructed, and a threshold is applied to create an adjacency matrix, which serves as the foundation for a functional network. Graph theory metrics, such as the clustering coefficient (measure of network segregation) and global efficiency (measure of network integration), can then be computed from this adjacency matrix to quantitatively describe the network's topology [14].
The Scientist's Toolkit: Essential Reagents and Materials
The execution of a standardized calcium imaging experiment relies on a core set of research reagents and tools. The following table details these essential components.
Table 2: Key Research Reagent Solutions for Calcium Imaging
| Category | Item | Function & Key Characteristics |
|---|---|---|
| Genetically Encoded Calcium Indicators (GECIs) | GCaMP Series (e.g., GCaMP6) [35] | Intensiometric, single-wavelength green indicator; high dynamic range; widely used for detecting neuronal activity. |
| NEMOer Series (e.g., NEMOer-f) [28] | ER/SR-targeted GECIs; large dynamic range (up to 80x greater than G-CEPIA1er); enables detection of elementary SR Ca²⁺ releases (e.g., Ca²⁺ blinks). | |
| G-Ca-FLITS [113] | Fluorescence Lifetime Imaging Microscopy (FLIM) biosensor; remains bright in both Ca²⁺-bound and unbound states; enables quantitative measurement of absolute Ca²⁺ concentration. | |
| Chemical Dyes | Fura-2 [4] [45] | Ratiometric dye (excitation at 340/380 nm); allows for quantitative calibration of intracellular [Ca²⁺]; used in agonist-induced signaling experiments. |
| Fluo-4 [35] | Intensiometric, single-wavelength green dye; high signal-to-noise ratio for detecting transient Ca²⁺ changes. | |
| Analysis Software & Packages | CalciumNetExploreR (CNER) [14] | R package for downstream analysis; performs normalization, binarization, network construction, and topology analysis in a unified pipeline. |
| Custom Jupyter Notebooks [4] | Python-based scripts for flexible, lab-specific analysis; common functions include ROIDetection, PeakDetection, and Frequency_Analysis. | |
| Specialized Microscopy | Light Beads Microscopy (LBM) [45] | Advanced volumetric imaging technique; acquisition speed is limited by fluorophore lifetime; benefits from indicators with shortened, Ca²⁺-insensitive lifetimes. |
| Multiphoton Microscopy [4] | Enables deep-tissue imaging with reduced phototoxicity; used for in vivo recordings in live animals. |
The field of calcium imaging stands at a critical juncture. The power of the technique to reveal the intricate dynamics of one of biology's most fundamental signaling systems is undeniable. However, the current lack of standardization, evidenced by the diverse and often incompatible analysis methods employed across studies, poses a significant threat to data reproducibility, comparative evaluation, and ultimately, scientific progress. The widespread adoption of common data sets and standardized analysis pipelines, such as the modular workflow and detailed methodologies outlined in this whitepaper, is an imperative next step. By embracing these tools and principles, the community of researchers and drug developers can transcend methodological inconsistencies, unlock the full potential of calcium imaging data, and collectively accelerate the deciphering of the complex calcium code.
Calcium ions (Ca²⁺) are ubiquitous intracellular messengers governing processes from neuronal communication and muscle contraction to gene expression and cell death. The ability to detect and image calcium dynamics with high spatiotemporal resolution is therefore fundamental to modern life science research and drug development. This whitepaper examines the convergence of three transformative technologies revolutionizing calcium signaling detection: synthetic biology for designing intelligent cellular circuits, near-infrared genetically encoded calcium indicators (NIR-GECIs) for deep-tissue imaging, and emerging ultrasensitive sensors for endoplasmic reticulum (ER) calcium dynamics. These innovations are enabling unprecedented insight into cellular physiology and opening new frontiers in therapeutic intervention.
Current calcium detection methods have evolved from early chemical chelators to sophisticated genetically encoded indicators. The field is now shifting from mere observation to predictive, multiparameter cellular interrogation. This evolution is critical for understanding complex diseases such as neurodegenerative disorders, where disrupted calcium homeostasis is a hallmark feature, and for developing targeted therapies that require precise modulation of specific signaling pathways.
Synthetic Biology Frameworks for Advanced Biosensing
Synthetic biology applies engineering principles to biological systems, creating standardized, predictable components for programming cellular behavior. This approach has moved from conceptual demonstrations to robust therapeutic platforms, particularly in cell-based therapies and advanced biosensing.
Engineering Therapeutic Cellular Circuits
The development of Chimeric Antigen Receptor (CAR)-T cells exemplifies the successful application of synthetic biology in medicine. CARs are synthetic receptors comprising an antigen-binding domain fused to T-cell activating signaling domains. The evolution through three generations has incorporated multiple co-stimulatory domains (e.g., CD28, 4-1BB) to enhance potency and persistence [114]. These engineered cells demonstrate the core synthetic biology principle of modular design: extracellular recognition domains can be swapped to target different antigens while maintaining conserved intracellular signaling architecture. Clinical successes include FDA-approved therapies such as Kymriah (tisagenlecleucel) for acute lymphoblastic leukemia and Yescarta (axicabtagene ciloleucel) for large B-cell lymphoma, with response rates exceeding 50% in some trials [114].
Beyond cancer, synthetic biology enables the creation of biosensor-based therapeutic tools that detect intracellular biomarkers and trigger therapeutic responses. These systems employ genetic circuits that sense disease states through specific molecular signatures—such as abnormal metabolite concentrations, pathological protein aggregates, or inflammatory cytokines—and respond with programmed actions including controlled drug release, apoptosis induction, or corrective gene expression [115]. The design-build-test-learn (DBTL) cycle, accelerated by CRISPR/Cas9 genome editing and low-cost DNA synthesis, has dramatically accelerated the development of these sophisticated cellular machines [114].
Biosensor Design Principles and Classification
Biosensors integrate a biological recognition element with a physicochemical transducer to detect and quantify specific analytes. Effective biosensors must balance sensitivity (ability to detect low analyte concentrations), selectivity (discrimination against interfering substances), and stability (consistent performance over time) [116].
Biosensors are traditionally classified by transducer type:
- Electrochemical biosensors measure currents from oxidation/reduction reactions
- Optical biosensors detect changes in light properties (intensity, wavelength)
- Piezoelectric biosensors measure mass changes through frequency shifts
- Thermal biosensors detect heat changes from biochemical reactions [116]
A novel classification based on substrate morphology (e.g., paper, electrospun fiber mats, thin films) is emerging, recognizing how material properties influence sensor performance through effects on surface area, diffusion kinetics, and bioreceptor immobilization [116].
Table 1: Biosensor Classification and Applications in Calcium Signaling Research
| Classification Basis | Sensor Type | Key Characteristics | Applications in Calcium Research |
|---|---|---|---|
| Transducer Mechanism | Electrochemical | Measures electrical current/voltage | Miniaturized sensors for extracellular Ca²⁺ |
| Optical | Detects light property changes | Genetically encoded Ca²⁺ indicators (GECIs) | |
| Piezoelectric | Sensitive to mass changes | Detection of Ca²⁺-binding events | |
| Bioreceptor Element | Enzyme-based | Catalytic signal amplification | Detecting Ca²⁺-activated enzymes |
| Antibody-based | High specificity | Immobilizing Ca²⁺-binding proteins | |
| Aptamer-based | Nucleic acid recognition | Conformational changes with Ca²⁺ binding | |
| Substrate Morphology | Paper-based | Low-cost, portable | Point-of-care calcium testing |
| Nanofiber-based | High surface area | Enhanced sensitivity Ca²⁺ detection |
Near-Infrared Genetically Encoded Calcium Indicators
Near-infrared GECIs represent a quantum leap in calcium imaging technology, addressing fundamental limitations of visible-light indicators including tissue autofluorescence, light scattering, and spectral incompatibility with optogenetic actuators.
Spectral Advantages and Engineering Challenges
The near-infrared optical window (650-900 nm) offers superior tissue penetration due to reduced light scattering and minimal absorption by hemoglobin, melanin, and other endogenous chromophores [117] [33]. Imaging in this window enables deeper tissue observation with lower background fluorescence and reduced phototoxicity. Additionally, NIR indicators eliminate spectral crosstalk when combined with most optogenetic tools and visible-light fluorescent proteins, enabling simultaneous manipulation and measurement of multiple cellular parameters [117].
Engineering effective NIR-GECIs presents unique challenges. Unlike green fluorescent protein (GFP)-based indicators, NIR indicators utilize biliverdin (BV) as a chromophore, an abundant product of heme catabolism in mammalian cells [117]. Successful NIR-GECI design must ensure efficient BV binding, high molecular brightness, and significant conformational changes upon calcium binding that translate to measurable fluorescence changes.
Current NIR-GECI Platforms: A Technical Comparison
Three major NIR-GECI platforms represent the state of the art, each with distinct advantages and limitations:
iGECI is a FRET-based indicator employing miRFP670 and miRFP720 as donor and acceptor fluorescent proteins, respectively, with a calmodulin-M13 calcium-sensing domain [117]. It exhibits a 600% increase in FRET/donor fluorescence ratio upon calcium binding, high brightness, and excellent photostability. However, its large size (86 kDa) presents challenges for viral packaging, and its response kinetics are relatively slow [118].
NIR-GECO2G is an intensiometric indicator based on a modified mIFP protein [118]. It offers faster on/off kinetics suitable for detecting single action potentials and higher response magnitude per single action potential (up to 17% ΔF/F) in neurons. However, it suffers from relatively low cellular brightness and rapid photobleaching [118].
FR-GECO1a/1c are recently developed far-red indicators based on mKelly fluorescent proteins with excitation/emission maxima at ~596/~644 nm [33]. FR-GECO1c exhibits an impressive 18-fold fluorescence increase upon calcium binding, high brightness under both one- and two-photon illumination, and affinities suitable for neuronal imaging (apparent Kd = 83 nM) [33].
Table 2: Performance Characteristics of Near-Infrared and Far-Red GECIs
| Indicator | Type | Ex/Emmax (nm) | Ca²⁺ Response (ΔF/F or ΔR/R) | Apparent Kd (nM) | Molecular Brightness | Key Applications |
|---|---|---|---|---|---|---|
| iGECI | FRET-based | 640/670, 700/720 | 600% ratio change | 15 and 890 (two affinities) | High | Deep-tissue imaging, hybrid photoacoustic imaging |
| NIR-GECO2G | Intensiometric | 678/704 | Up to 17% per action potential | Not specified | Low | Fast neuronal activity detection |
| FR-GECO1a | Intensiometric | 596/642 | 6-fold increase | 29 | High | All-optical manipulation and measurement |
| FR-GECO1c | Intensiometric | 596/646 | 18-fold increase | 83 | High | High-contrast neuronal imaging |
| iGECInano | FRET-based | Not specified | 60% donor/FRET ratio drop | Not specified | High (4-6× iGECI in bacteria) | Neuronal imaging with improved SNR and kinetics |
Experimental Protocol: Validating NIR-GECI Performance in Neurons
Objective: Characterize NIR-GECI performance in detecting spontaneous and evoked neuronal activity.
Materials:
- Primary mouse cortical/hippocampal neurons (DIV 14-21)
- NIR-GECI plasmid (iGECI, FR-GECO1c, or iGECInano) in mammalian expression vector
- Lipofectamine 3000 or viral delivery system (AAV, lentivirus)
- Imaging solution: 125 mM NaCl, 5 mM KCl, 2 mM CaCl₂, 1 mM MgCl₂, 10 mM HEPES, 10 mM glucose, pH 7.4
- One-photon or two-photon microscope with appropriate NIR lasers and detectors
- Field stimulation electrode or optogenetic setup for evoked activity
- Pharmacological agents: TTX (1 μM) for blocking action potentials, KCl (50 mM) for depolarization
Methodology:
- Neuronal Transfection/Transduction: Introduce NIR-GECI construct into neurons using calcium phosphate transfection (plasmids) or viral transduction (AAV). Allow 5-7 days for expression and indicator maturation.
- Microscopy Setup: Configure imaging system with appropriate excitation (e.g., 640 nm for iGECI, 596 nm for FR-GECOs) and emission filters (670/720 nm for iGECI, 640/700 nm for FR-GECOs).
- Baseline Imaging: Acquire time-series images at 5-20 Hz frame rate under baseline conditions for 2-5 minutes to detect spontaneous activity.
- Evoked Activity: Apply field stimulation (1 ms pulses, 10-20 Hz, 2s duration) or optogenetic activation (for co-expressed actuators) while imaging.
- Pharmacological Validation: Apply KCl (50 mM) to induce sustained depolarization and calcium influx, or TTX (1 μM) to block action potentials and confirm specificity.
- Data Analysis: Calculate ΔF/F or ΔR/R for regions of interest corresponding to neuronal somata or processes. Detect calcium transients using peak detection algorithms and correlate with stimulation parameters.
Troubleshooting: Low signal-to-noise may require optimization of expression time, BV supplementation (100 μM for 4-24 hours), or laser power adjustment. Slow kinetics may indicate insufficient indicator maturation or suboptimal temperature maintenance (37°C) [117] [33] [118].
Ultrasensitive ER Calcium Sensing
The endoplasmic reticulum serves as the major intracellular calcium store, with luminal calcium concentration ([Ca²⁺]ER) dynamically regulated by IP₃ receptors, ryanodine receptors, and SERCA pumps. Dysregulated ER calcium homeostasis is implicated in numerous pathologies including neurodegenerative diseases, making accurate ER calcium measurement essential.
Calcium Signaling Pathways in Cellular Communication
Calcium signaling operates across multiple spatial and temporal scales, from localized microdomains to global waves, and from millisecond transients to sustained oscillations. In neural systems, calcium mediates bidirectional communication between neurons and astrocytes, with both intracellular and extracellular calcium pools contributing to signal integration [9].
Intracellular calcium elevations in astrocytes originate from multiple sources including ER release via IP₃ receptors, store-operated calcium entry (SOCE) through STIM/Orai complexes, reverse operation of the Na⁺-Ca²⁺ exchanger (NCX), and various ionotropic and metabotropic receptors [9]. These signals regulate critical processes such as gliotransmitter release, metabolic support, and ion homeostasis.
Emerging evidence reveals that extracellular calcium ([Ca²⁺]o) is not merely a passive reservoir but an active signaling mediator that influences neuronal excitability on millisecond timescales through mechanisms including calcium-sensing receptor (CaSR) activation, ion channel modulation, and ephaptic coupling [9]. This expanded view of calcium signaling necessitates sensors capable of resolving dynamics in multiple cellular compartments simultaneously.
Advanced ER Sensor Design Considerations
Engineering effective ER calcium sensors presents unique challenges distinct from cytosolic indicators. Ideal ER sensors must:
- Tolerate the unique ER luminal environment (high [Ca²⁺], oxidizing conditions, specific chaperones)
- Exhibit appropriate affinity and dynamic range for the high resting [Ca²⁺]ER (~100-800 μM)
- Incorporate ER retention signals (e.g., KDEL sequences) while maintaining proper folding
- Minimally perturb native calcium handling by endogenous buffers and pumps
Recent advances include targeted versions of existing GECIs with modified calcium affinities and ER-optimized folding, as well as entirely new scaffolds designed specifically for the ER environment. These sensors are revealing the spatiotemporal complexity of ER calcium dynamics during processes such as store-operated calcium entry, ER-mitochondrial calcium transfer, and ER stress responses.
Integrated Experimental Approaches and Future Directions
The convergence of synthetic biology, advanced imaging, and computational analysis is creating unprecedented opportunities for deciphering complex calcium signaling networks.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Advanced Calcium Signaling Studies
| Reagent Category | Specific Examples | Function/Application |
|---|---|---|
| NIR-GECIs | iGECI, FR-GECO1c, iGECInano | Deep-tissue calcium imaging with minimal spectral crosstalk |
| Optogenetic Actuators | Channelrhodopsins (ChR2), NIR opsins | Optical control of neuronal activity compatible with NIR imaging |
| Calcium Modulators | Ionomycin, Thapsigargin, EGTA | Experimental manipulation of calcium levels for sensor validation |
| BV Chromophore | Biliverdin HCl | Supplemental chromophore for improved NIR-FP brightness |
| Viral Delivery Systems | AAV, Lentivirus | Efficient neuronal transduction for stable sensor expression |
| Synthetic Biology Parts | Promoters, CAR domains, Genetic circuits | Programming cellular responses to calcium signals |
Emerging Applications and Methodological Convergence
The integration of live imaging with synthetic biology is enabling new classes of experiments where observation and intervention occur simultaneously in intact systems. Live imaging provides direct observation of dynamic biological processes across scales, capturing progression from molecular to organismal levels with spatiotemporal resolution [119]. When combined with synthetic circuits that respond to specific calcium signatures, researchers can move beyond correlation to establish causality in signaling pathways.
Future directions include:
- Multiparameter imaging systems combining NIR-GECIs with neurotransmitter, metabolic, and voltage indicators
- Closed-loop synthetic circuits that detect pathological calcium signatures and trigger corrective interventions
- Nanoparticle-enhanced delivery of calcium modulators to specific cellular compartments
- AI-assisted analysis of complex calcium signaling patterns across cell populations
These technological advances will continue to dissolve boundaries between observation and intervention, ultimately enabling precise spatiotemporal control of calcium signaling for both basic research and therapeutic applications.
The integration of synthetic biology principles with advanced NIR indicators and compartment-specific sensors represents a paradigm shift in calcium signaling research. These technologies are transforming our ability to observe and manipulate cellular communication in real-time within complex physiological environments. For researchers and drug development professionals, these tools offer unprecedented resolution for decoding disease mechanisms and validating therapeutic interventions that target calcium signaling pathways. As these technologies mature and converge, they will undoubtedly illuminate new aspects of cellular signaling and enable innovative approaches to treating diseases characterized by calcium dysregulation.
Conclusion
The field of calcium detection has matured into a sophisticated discipline, offering researchers an unprecedented window into cellular activity. The ongoing development of indicators, such as the ultra-fast jGCaMP8 for neural populations and the highly sensitive NEMOer for ER calcium, continues to push the boundaries of what is observable. However, the power of these tools is fully realized only through rigorous validation, standardized analysis, and a clear understanding of their inherent trade-offs. Future progress hinges on interdisciplinary efforts combining synthetic biology for novel sensor design, advanced computational models for precise kinetic analysis, and the development of standardized validation frameworks. These advances will not only deepen our fundamental understanding of calcium's 'code' in health and disease but will also accelerate drug discovery by providing more precise, high-throughput functional readouts for therapeutic screening and mechanistic studies.
References
- 1. Calcium Ions as Intracellular Messengers [https://link.springer.com/chapter/10.1007/978-1-59259-032-2_9]
- 2. Calcium Signalling: More Messengers, More Channels, ... [https://www.sciencedirect.com/science/article/pii/S0960982202010552]
- 3. Simultaneous detection of dynamic calcium signaling and ... [https://www.biophysics-reports.org/en/article/doi/10.52601/bpr.2023.230038]
- 4. Tools for Quantitative Analysis of Calcium Signaling Data ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10312663/]
- 5. Calcium, an Emerging Intracellular Messenger for the ... [https://pubmed.ncbi.nlm.nih.gov/34268313/]
- 6. Fast and sensitive GCaMP calcium indicators for imaging ... [https://www.nature.com/articles/s41586-023-05828-9]
- 7. Calcium, an Emerging Intracellular Messenger for the ... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2021.694828/full]
- 8. FRET Imaging of Calcium Signaling in Live Cells under ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4165894/]
- 9. Calcium Dynamics in Astrocyte-Neuron ... - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC12610994/]
- 10. Rapid detection of neurons in widefield calcium imaging ... [https://www.nature.com/articles/s41592-023-01838-7]
- 11. Modeling traveling calcium waves in cellular structures [https://link.springer.com/article/10.1007/s10827-025-00898-2]
- 12. Lessons from the use of in vivo cellular calcium imaging ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12605308/]
- 13. CalTrig: A GUI-Based Machine Learning Approach for ... [https://www.eneuro.org/content/12/7/ENEURO.0009-25.2025]
- 14. an R package for network analysis of calcium imaging data [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-025-06206-0]
- 15. an R package for network analysis of calcium imaging data [https://pubmed.ncbi.nlm.nih.gov/40859128/]
- 16. Comprehensive detection and characterization of neuronal ... [https://www.sciencedirect.com/science/article/pii/S2352711025004017]
- 17. Fast, accurate, and versatile data analysis platform for the ... [https://www.sciencedirect.com/science/article/pii/S0092867425002855]
- 18. Imaging calcium signals in vivo: a powerful tool in physiology ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3166690/]
- 19. Aequorin-based measurements of intracellular Ca2+-signatures in plant cells - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC145563/]
- 20. An Automated Aequorin Luminescence-Based Functional Calcium Assay for G-Protein-Coupled Receptors - ScienceDirect [https://www.sciencedirect.com/science/article/abs/pii/S0003269799941453]
- 21. Calcium & Other Ion Indicators [https://biotium.com/technology/cellular-stains/ion-indicators-membrane-potential-dyes/]
- 22. Calcium Indicators | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/life-science/cell-analysis/cell-viability-and-regulation/ion-indicators/calcium-indicators.html]
- 23. Far-red fluorescent genetically encoded calcium ion ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11977202/]
- 24. Establishment of MHHi001-A-5, a GCaMP6f and RedStar ... [https://www.sciencedirect.com/science/article/pii/S1873506121000520]
- 25. Fast Kinetics of Calcium Signaling and Sensor Design - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4553074/]
- 26. Calcium imaging [https://en.wikipedia.org/wiki/Calcium_imaging]
- 27. How Does Calcium Imaging Work | Calcium Indicators [https://andor.oxinst.com/learning/view/article/an-overview-of-calcium-imaging-calcium-indicators-and-experimental-considerations]
- 28. Highly dynamic and sensitive NEMOer calcium indicators ... [https://www.nature.com/articles/s41467-025-58705-6]
- 29. Protocol for live-cell calcium imaging of human lung ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12451350/]
- 30. Live Cell Calcium Indicators [https://www.sigmaaldrich.com/US/en/technical-documents/technical-article/cell-culture-and-cell-culture-analysis/imaging-analysis-and-live-cell-imaging/calcium-indicators?srsltid=AfmBOoousGEAtTbHQ7CzINGqn_01sJlNn04GNn5ZPrYT1CVRIyv1GqQu]
- 31. How to Use Calcium Imaging for Investigating Cell Signaling [https://ibidi.com/content/917-how-to-use-calcium-imaging-for-investigating-cell-signaling]
- 32. Fluorescent and Bioluminescent Calcium Indicators with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9026248/]
- 33. Far-red fluorescent genetically encoded calcium ion ... [https://www.nature.com/articles/s41467-025-58485-z]
- 34. Probing neuronal activity with genetically encoded calcium ... [https://www.sciencedirect.com/science/article/pii/S0168010224000762]
- 35. A Review of Calcium Activity Analysis Methods Employed to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10813340/]
- 36. Live-Cell Imaging | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/life-science/cell-analysis/cellular-imaging/fluorescence-microscopy-and-immunofluorescence-if/microscopy-reagents-and-media/live-cell-imaging-reagents.html]
- 37. GaN photonic microchip for label-free monitoring of single ... [https://www.sciencedirect.com/science/article/abs/pii/S2666998625001243]
- 38. Live Cell Calcium Indicators [https://www.sigmaaldrich.com/US/en/technical-documents/technical-article/cell-culture-and-cell-culture-analysis/imaging-analysis-and-live-cell-imaging/calcium-indicators?srsltid=AfmBOoqKyhxvkeH6yX-4ddCUeY4sM1dkqye89LwxnJuUCs01PDfqoS3n]
- 39. Fura-2 AM imaging protocol [https://www.abcam.com/en-us/technical-resources/protocols/fura-2-am-imaging]
- 40. Imaging and analysis of genetically encoded calcium ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6609268/]
- 41. Calcium imaging: Unraveling the neurobiological ... [https://www.sciencedirect.com/science/article/abs/pii/S0143416025000636]
- 42. High-throughput-compatible assays using a genetically- ... [https://www.nature.com/articles/s41598-019-49070-8]
- 43. Highly dynamic and sensitive NEMOer calcium indicators for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11992197/]
- 44. A highly sensitive fluorescent indicator dye for calcium ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4232931/]
- 45. Calcium Indicators with Fluorescence Lifetime-Based ... [https://www.mdpi.com/1422-0067/25/23/12493]
- 46. Calcium imaging: a technique to monitor calcium dynamics in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10784449/]
- 47. Transfection Technologies for Next-Generation Therapies - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12347970/]
- 48. Efficient transduction of pancreas tissue slices with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11974670/]
- 49. An updated suite of viral vectors for in vivo calcium ... [https://www.nature.com/articles/s41467-023-36324-3]
- 50. Rapid Optimization of Gene Delivery by Parallel End- ... [https://pubmed.ncbi.nlm.nih.gov/28182918/]
- 51. Fluorescent sensors for imaging of interstitial calcium [https://www.nature.com/articles/s41467-023-41928-w]
- 52. Engineered nucleocytosolic vehicles for loading of ... [https://www.sciencedirect.com/science/article/pii/S0092867425002880]
- 53. The Many Flavors of Widefield Microscopy [https://bitesizebio.com/19409/the-many-flavors-of-widefield-microscopy/]
- 54. Introduction to Confocal Microscopy [https://evidentscientific.com/en/microscope-resource/knowledge-hub/techniques/confocal/confocalintro]
- 55. Confocal Microscopy [https://www.bostonind.com/blog/confocal-microscopy?srsltid=AfmBOoqVGRbNPGrQvf6LNGQYj4bA05QJfsmhFhteJfimT-neeTgX0i8D]
- 56. Diving into the deep: confocal vs multi-photon microscopy [https://abberior.rocks/knowledge-base/diving-into-the-deep-confocal-vs-multi-photon-microscopy/]
- 57. Concepts in Confocal Microscopy [https://evidentscientific.com/en/microscope-resource/knowledge-hub/techniques/confocal]
- 58. More than double the fun with two-photon excitation ... [https://www.nature.com/articles/s42003-024-06057-0]
- 59. What are the advantages and disadvantages of a confocal ... [https://szphoton.com/blogs/articles/what-are-the-advantages-and-disadvantages-of-a-confocal-microscope]
- 60. Depth-variant deconvolution applied to widefield ... [https://www.nature.com/articles/s42003-025-08992-y]
- 61. An extension module for a two-photon microscope enables ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12506568/]
- 62. Calcium Imaging in Vivo: How to Correctly Select and Apply ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11980459/]
- 63. in neuroscience research: principles, applications... Fiber photometry [https://pmc.ncbi.nlm.nih.gov/articles/PMC11582208/]
- 64. GuPPy, a Python toolbox for the analysis of fiber data photometry [https://www.nature.com/articles/s41598-021-03626-9?error=cookies_not_supported&code=9270a85a-0103-4577-8ef5-7ccfdb13b526]
- 65. In Vivo Optical Microendoscopy for Imaging Cells Lying ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5731463/]
- 66. In vivo minimally invasive interstitial multi-functional ... [https://www.nature.com/articles/srep01805]
- 67. Chronic Ca2+ imaging of cortical neurons with long-term ... [https://elifesciences.org/articles/76691]
- 68. Mitochondrial or Cytosolic Live Using... Calcium Imaging [https://bio-protocol.org/en/bpdetail?id=3504&type=0]
- 69. Calcium signal compartmentalization [https://pubmed.ncbi.nlm.nih.gov/12415734/]
- 70. The ER mitochondria calcium cycle and ER stress response ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4039088/]
- 71. Indicators Live Cell Calcium [https://www.sigmaaldrich.com/US/en/technical-documents/technical-article/cell-culture-and-cell-culture-analysis/imaging-analysis-and-live-cell-imaging/calcium-indicators]
- 72. - Live in 3D Intestinal Organoids Cell Calcium Imaging [https://pubmed.ncbi.nlm.nih.gov/39395108/]
- 73. Calcium imaging analysis – how far have we come? - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8406438/]
- 74. From form to function: calcium compartmentalization in ... [https://www.nature.com/articles/nn0700_653]
- 75. Rules of life at the interface of calcium signaling and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12659936/]
- 76. Calcium assays: at the centre of biology [https://www.bmglabtech.com/en/blog/calcium-assays-at-the-centre-of-biology/]
- 77. Decoding Molecular Network Dynamics in Cells: Advances ... [https://www.mdpi.com/2079-6374/15/9/614]
- 78. Calcium imaging in retinal research: challenges and ... [https://www.aimspress.com/article/id/69153e64ba35de34708ade17]
- 79. An expanded palette of bright and photostable organellar ... [https://elifesciences.org/reviewed-preprints/107845]
- 80. Keeping Cells Alive in Microscopy [https://www.mdpi.com/2673-4125/5/1/1]
- 81. Potential consequences of phototoxicity on cell function ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11567556/]
- 82. A quantitative hypermorphic CNGC allele confers ectopic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5716663/]
- 83. Illuminating calcium and potassium dynamics with red ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12551949/]
- 84. A Multicellular analysis Calcium Imaging toolbox for ImageJ [https://pubmed.ncbi.nlm.nih.gov/40894801/]
- 85. A comprehensive suite for extracting neuron signals across ... [https://www.nature.com/articles/s41467-025-58817-z]
- 86. Minian, an open-source miniscope analysis pipeline [https://elifesciences.org/articles/70661]
- 87. A Preprocessing Toolbox for 2-Photon Subcellular Calcium ... [https://www.eneuro.org/content/12/5/ENEURO.0565-24.2025]
- 88. SpecSeg is a versatile toolbox that segments neurons and ... [https://www.sciencedirect.com/science/article/pii/S2667237522001813]
- 89. Quantitative analysis of miniature synaptic calcium transients ... [https://pubs.rsc.org/en/content/articlehtml/2025/dd/d4dd00197d]
- 90. SICT: automated detection and supervised inspection of ... [https://www.nature.com/articles/s41598-018-33847-4]
- 91. CalTrig: A GUI-Based Machine Learning Approach for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12263099/]
- 92. Calcium transient detection and SWR coincidence analysis [https://bio-protocol.org/exchange/minidetail?id=8359165&type=30]
- 93. A Review of Calcium Activity Analysis Methods Employed ... [https://www.mdpi.com/2218-273X/14/1/138]
- 94. How to compute ΔF/F from calcium imaging data |… - Scientifica [https://www.scientifica.uk.com/learning-zone/how-to-compute-%CE%B4f-f-from-calcium-imaging-data]
- 95. A simple method for quantitative calcium imaging in ... [https://www.sciencedirect.com/science/article/abs/pii/S0165027009004373]
- 96. Calibration Protocol for Fluorescent Calcium Indicators [https://www.aatbio.com/resources/application-notes/calibration-protocol-for-fluorescent-calcium-indicators]
- 97. Ca2+ imaging of synaptic compartments using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10820801/]
- 98. Optimizing Calcium Detection Methods in Animal Systems [https://pmc.ncbi.nlm.nih.gov/articles/PMC7996158/]
- 99. A comparison of neuronal population dynamics measured ... [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1008198]
- 100. GCaMP - Wikipedia [https://en.wikipedia.org/wiki/GCaMP]
- 101. Engineering of NEMO as calcium indicators with large ... [https://www.nature.com/articles/s41592-023-01852-9]
- 102. GCaMP [https://grokipedia.com/page/GCaMP]
- 103. Fast and sensitive GCaMP calcium indicators for imaging ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10060165/]
- 104. Janelia scientists develop fastest calcium indicators yet | Janelia Research Campus [https://www.janelia.org/news/janelia-scientists-develop-fastest-calcium-indicators-yet]
- 105. jGCaMP8 Calcium indicators | Janelia Research Campus [https://www.janelia.org/jgcamp8-calcium-indicators]
- 106. jGCaMP8 Fast Genetically Encoded Calcium Indicators [https://janelia.figshare.com/articles/online_resource/jGCaMP8_Fast_Genetically_Encoded_Calcium_Indicators/13148243]
- 107. Development and characterization of novel jGCaMP8f ... [https://www.frontiersin.org/journals/cellular-neuroscience/articles/10.3389/fncel.2023.1155406/full]
- 108. Fitting and comparison of calcium-calmodulin kinetic schemes ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0318646]
- 109. Analysis of calcium signaling in live human Tongue cell 3D ... [https://pubmed.ncbi.nlm.nih.gov/32014795/]
- 110. A programmable microfluidic platform to monitor calcium ... [https://www.nature.com/articles/s41378-024-00733-1]
- 111. Tracking calcium dynamics from individual neurons in ... [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1009432]
- 112. Analysis pipelines for calcium imaging data [https://www.sciencedirect.com/science/article/pii/S0959438818300941]
- 113. A green lifetime biosensor for calcium that remains bright ... [https://elifesciences.org/reviewed-preprints/105086]
- 114. Applications of synthetic biology in medical and ... [https://www.nature.com/articles/s41392-023-01440-5]
- 115. Biosensor-based therapy powered by synthetic biology [https://www.sciencedirect.com/science/article/pii/S2590183422000515]
- 116. Sensing the Future—Frontiers in Biosensors: Exploring Classifications, Principles, and Recent Advances - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11656264/]
- 117. A near-infrared genetically encoded calcium indicator for in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7956128/]
- 118. Design and Initial Characterization of a Small Near-Infrared ... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2022.880107/full]
- 119. To see and to know: the power of live imaging ... [https://www.sciencedirect.com/science/article/pii/S1673852725002826]